TidyGWAS
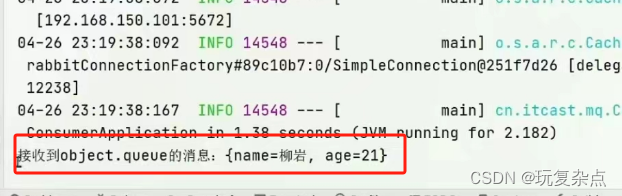
GWAS分析关键结果之一是显著性SNP位点的P值,通常多年份多地点多模型的GWAS分析将会产生很多结果文件,如何对这些数据进行整理?
汇总这些结果,并将显著性的位点或区域找出来,更加清晰的展示关键信息。

今天介绍TidyGWAS结果整理新方法,前段时间曾发过一篇笔记(GWAS结果整理算法),但是有一些地方比较繁琐,仍有优化空间。
本篇笔记将分享一种基于R语言自动实现GWAS结果整理的升级版方法,通过优化关键步骤和算法,将代码量从2000行缩减到了400行,速度提高10倍以上。
关键词:GWAS、R语言、Tidyverse
前期准备工作
软件安装
本次使用的R语言版本是R4.3.0,需要以下R包,如果没有安装需要提前安装。
# Install packages
install.packages("tidyverse")
install.packages("data.table")
install.packages("foreach")
install.packages("doParallel")
install.packages("stringr")# Load libraries
library(tidyverse)
library(data.table)
library(foreach)
library(doParallel)
library(stringr)
项目文件
通常建议每个任务建立项目文件夹,请新建一个文件夹并设置为工作目录,然后创建如下文件结构。
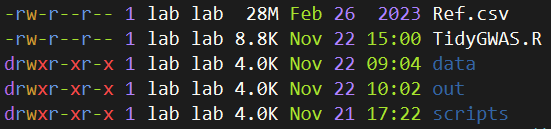
其中Ref是参考基因组信息,data子文件夹存放原始的数据,可以包含很多个以txt结尾的文件,该文件内容如下所示:
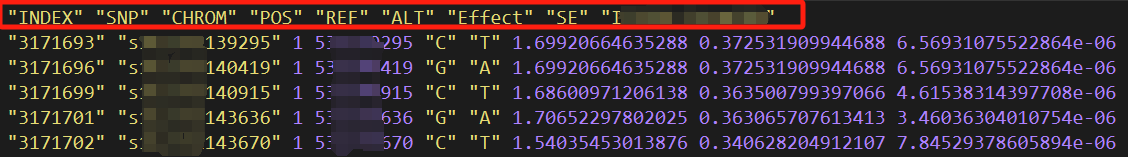
主要信息是SNP、染色体、物理位置、显著性P值。每个文件的命名方式是“类型.年份环境.模型.P阈值.txt”
核心操作步骤
人工设置参数
# 参数设置-----
prefix <- "xxx"
windows_near <- 300*1000
#默认300kb内为连续显著区段
id_list <- list.files("./data/",pattern = "*.txt") # 待整理的所有文件
Ref <- fread("./Ref.csv") # 参考基因组信息
其实,使用起来非常简单,只用设置一个项目输出结果前缀和窗口大小即可,这个窗口大小指的是在判定连续型显著区域时最大的阈值。
比如默认300Kb,假如某几个显著的SNP位点之间的物理距离都在300Kb之内,则把它们当做一个连续的显著区段。
之后的步骤都是全自动执行,不用再进行任何修改,如果您想使用此方法只需要直接运行一遍全部代码即可。
自动生成参数
id_df <- str_split(id_list,"[.]") %>% as.data.frame() %>% t() %>% as.data.frame()
type_list <- id_df$V1[!duplicated(id_df$V1)]
phe_list <- id_df$V2[!duplicated(id_df$V2)]
model_list <- id_df$V3[!duplicated(id_df$V3)]
p_value <- 1e-5 #显著性
suffix <- ".txt"
write.csv(id_df,str_c(prefix,"_parment.csv"),row.names = F)
这段代码的目的是将一个包含点号分隔字符串的列表(文件名称列表)分割成多个部分,转换为数据框,然后从每一列中提取出不重复的元素,分别存储在三个不同的列表中,这样就得到了所有待整理的信息清单。
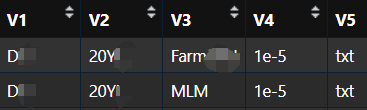
这份文件将会自动输出保存,记录了所有待整理的文件信息和参数。
创建输出文件夹
out_dirs <- c("1_GWAS_Result_txt2csv","2_SNP_Infomation","3_Gene_Maping_Result","4_Rebind_All_Output")
for (mydir in out_dirs){if (dir.exists(paste0("./out/",mydir))){cat(paste0("[check] ./out/",mydir," is exist !\n"))}else{dir.create(paste0("./out/",mydir))cat(paste0("[check] ./out/",mydir," create finished !\n"))}
}
这一步自动检测是否存在目标文件夹,如果不存在的话创建一个,后续的中间结果和文件将自动写入这些文件夹。
Step1:文件整理
for (id in id_list){file_name <- paste0("./data/",id)atom <- str_split(id,"[.]")type <- atom[[1]][1]phe <- atom[[1]][2]way <- atom[[1]][3] %>% str_replace("Farm","") # 将模型替换为CPUplast <- atom[[1]][4]# 特异性标注P值并将其装换为数字型if (plast == "1e-5"){plast <- 6}else{plast <- as.numeric(plast)}print(file_name)# 计算p值并筛选df <- read_delim(file_name,delim = " ",col_types = cols(CHROM = col_character()))colnames(df)[9] <- waydf %>% mutate(log = round(-log10(!!sym(way)),1)) %>% filter(log > plast) ->data# 转换染色体编号i <- 1new <- data.frame(matrix(ncol = 2))new <- new[-1,]for (x in c(1:7)){for (y in c("A","B","D")){chr <- paste0(x,y)# print(chr)new_add <- c(i,chr)new <- rbind(new,new_add)i <- i + 1}}colnames(new) <- c("CHROM","chr")# 替换染色体编号data %>% left_join(new,by = "CHROM") ->data2data2$loc <- phe# 待标注的log值筛选data2$logwt <- ifelse(data2$log > 10,paste0('log=',data2$log,sep=""),NA)data2$MB <- data2$POS/1000000# 写出为中间结果write_csv(data2,paste0("./out/1_GWAS_Result_txt2csv/",type,".",phe,".",way,".csv"))
}
这段代码主要用于处理基因组数据,涉及文件读取、数据分割、条件筛选、数据转换和导出等步骤。
Step2:统计显著位点
Ref_chr <- Ref
all_single <- list() #汇总单标记
all_near <- list() #汇总连续标记
id_list_step2 <- list.files("./out/1_GWAS_Result_txt2csv/")
for (id in id_list_step2){# 创建染色体chr_list <- list()for (tmp_chr in new$chr){chr_list[[tmp_chr]] <- filter(Ref_chr,chr == tmp_chr)}# 开始计算----file_name <- paste0("./out/1_GWAS_Result_txt2csv/",id)atom <- str_split(id,"[.]")# print(file_name)data <- read_csv(file_name,show_col_types = FALSE)loc <- data$loc[1] #job <- paste0(atom[[1]][1],"_",atom[[1]][2],"_",atom[[1]][3])way <- atom[[1]][3]### 单标记筛选 ========================================================================# 计算基因位置间距data$longH <- NAdata$longQ <- NAdata$class <- NA# 显著位点小于3个的情况下跳过if (nrow(data) < 3){next}for (i in 2:nrow(data)){a <- data$POS[i]i <- i+1b <- data$POS[i]i <- i-2c <- data$POS[i]i <- i+1if(i == nrow(data)){data$class[i] <- "wei"break}if(a-c<0){data$class[i] <- "shou"next}if(b-a<0){data$class[i] <- "wei"next}data$longH[i] <- (b-a)data$longQ[i] <- (a-c)}data$class[1] <- "shou"# 对距离进行区分,按照windows_near为区分阈值for (i in 1:nrow(data)){if (is.na(data$longH[i]) | is.na(data$longQ[i])){next}if (data$longH[i]>windows_near & data$longQ[i]>windows_near){data$class[i] <- "single"}}# 单标记位点处理data$ws <- ifelse(is.na(data$logwt),paste0(data$SNP,",Find in ",str_replace(id,".csv",""),sep=""),paste0(data$SNP,",Find in ",str_replace(id,".csv",""),",",data$logwt,sep=""))### 单标记信息位置注释 ===================================================================# 单标记位置信息写入singlesingle <- data.frame(matrix(ncol = 4))single <- single[-1,]colnames(single) <- c("positon","info","chr","loc") for (i in 1:nrow(data)){tem_class <- data$class[i]tem_add <- c(data$POS[i],data$ws[i],data$chr[i],data$loc[i])ifelse(tem_class == 'single',single <- rbind(single,tem_add),"1")ifelse(tem_class == 'shou',single <- rbind(single,tem_add),"2")ifelse(tem_class == 'wei',single <- rbind(single,tem_add),"3")}colnames(single) <- c("positon","info","chr","loc") ### 连续区间筛选 ===================================================================near <- data.frame(matrix(ncol = 5)) #初始化矩阵near <- near[-1,]colnames(near) <- c("p1","p2","info","chr","number") for (x in c(1:7)){for (y in c("A","B","D")){chr_id <- paste0(x,y,sep="")foot <- 0 # 步长,用于迭代计算阅读框for (i in which(data$chr==chr_id)){if (sum(data$chr==chr_id)<2){next} #若某个染色体的位点数小于3则跳过if (foot>0){ # 如果foot变量大于0,说明两个位点存在跨越关系,进行归零foot <- foot - 1 # next}n_pos_1 <- data$POS[i]for (m in which(data$chr==chr_id)){n_pos_2 <- data$POS[m]if (n_pos_2 - n_pos_1 < 0){ # 后一个值小于前一个值时跳过next}else{if (n_pos_2 - n_pos_1 == 0){ # 两个值相等时跳过next}else{if (n_pos_2 - n_pos_1 < windows_near){ # 任意两个位点距离小于预设窗口大小foot <- foot+1 # 向前进行一步,跨越一个位点if (is.na(data$class[i])){data$class[i] <- "near" # 如果此时位点尚不属于single、shou、wei,则为near}}else{if (foot > 10){n_wn <- paste0(data$SNP[i],"-",data$SNP[m-1],", [",foot,"],Find in ",job)}else{n_wn <- paste0(data$SNP[i],"-",data$SNP[m-1],",Find in ",job)}n_add <- c(n_pos_1,data$POS[m-1],n_wn,data$chr[i],foot)if (is.na(data$class[i])){data$class[i] <- "near"}break}}}}if (length(data$POS[m-1])>0){if (n_pos_1 !=data$POS[m-1]){near <- rbind(near,n_add)n_add <- c()}}}}}colnames(near) <- c("p1","p2","info","chr","number")# 删除重复行near_new <- data.frame(matrix(ncol =5)) near_new <- near_new[-1,]for (i in 1:nrow(near)){if (!identical(near$p1[i],near$p2[i])){new_add <- c(near$p1[i],near$p2[i],near$info[i],near$chr[i],near$number[i])near_new <- rbind(near_new,new_add)}}colnames(near_new) <- c("p1","p2","info","chr","number") near <- near_new### 连续标记回帖参考基因组----OK <- 0 #成功添加的个数for (chr in names(chr_list)){for (i in 1:nrow(near)){if (identical(near$chr[i],chr)){my_a <- which.min(abs(as.numeric(near$p1[i]) - as.numeric(chr_list[[chr]]$X3G.Start.1)))my_b <- which.min(abs(as.numeric(near$p2[i]) - as.numeric(chr_list[[chr]]$X3G.Start.1)))chr_list[[chr]]$out[my_a:my_b] <- near$info[i]OK <- OK + (my_b - my_a)}}cli::cli_alert_success(str_c("[Chromosomes are currently being processed]:",chr))}num_near <- OK### 迭代添加单标记信息----OK <- 0 #成功添加的个数for (chr in names(chr_list)){for (i in 1:nrow(single)){if (identical(single$chr[i],chr)){index <- which.min(abs(as.numeric(single$positon[i]) - as.numeric(chr_list[[chr]]$X3G.Start.1)))chr_list[[chr]]$out[index] <- single$info[i]OK <- OK+1}}cli::cli_alert_success(str_c("[Chromosomes are currently being processed]:",chr))}num_single <- OK### 迭代添加首尾标记 =====================================================================cat(paste0("[",str_sub(data$ws[1],nchar(data$ws[1])-2,nchar(data$ws[1])),"-",data$loc[1],"] \t total near mark: ",num_near," \t total single mark: ",num_single,"\n"))all_near[[id]] <- nearall_single[[id]] <- singlewrite_excel_csv(single,paste0("./out/2_SNP_Infomation/",id,"_single.csv"))write_excel_csv(near,paste0("./out/2_SNP_Infomation/",id,"_near.csv"))write_excel_csv(data,paste0("./out/2_SNP_Infomation/",id,"_data.csv"),na = "near")out <- do.call(rbind,lapply(chr_list,function(x)x))write_excel_csv(out,paste0("./out/3_Gene_Maping_Result/",job,".snp.csv"),na = "")
}
这段R语言代码执行了一系列复杂的数据处理操作,主要用于处理基因组关联研究(GWAS)中的SNP(单核苷酸多态性)数据,包括识别显著的单个位点和连续区间,以及将这些信息映射到参考基因组上,这对于理解基因与特定表型之间的关系非常重要。
Step3:标注
id_list_step3 <- list.files("./out/3_Gene_Maping_Result/")
tem <- Ref[1:nrow(out),1:7] # 需要格外注意:tem和out文件必须一一对应
index <- 8 #从第8列开始标注
# 提取并标注注释信息
for (id in id_list_step3){now <- read.csv(paste0("./out/3_Gene_Maping_Result/",id))atom <- str_split(id,"[.]")job <- atom[[1]][1]tem <- bind_cols(tem,now[,8])colnames(tem)[index] <- jobindex <- index + 1# print(id)
}# 将多列信息合并为一列(优化算法)
tem <- tem %>% as.data.frame()
tem$all <- NA
tem_rm_na <- tem[,colSums(is.na(tem)) < nrow(tem)]
tem_rm_na_info <- tem_rm_na[,9:ncol(tem_rm_na)-1]
tem_rm_na$all <- apply(tem_rm_na_info, 1, function(x){x <- na.omit(x) # 删除NA值x <- x[nchar(x) > 3] # 保留字符长度大于3的元素paste(x, collapse = " ; ") # 使用分号作为分隔符连接字符串}
)
这段代码的主要目的是将多个基因组映射结果文件中的注释信息提取出来,并合并到一个主数据框中。每个文件的特定列(通常是第8列)被提取并添加到tem数据框中,最后将这些信息合并,以便进一步的分析和解释。
结果保存与输出
final_out <- cbind(tem_rm_na[,1:7],tem_rm_na$all,tem_rm_na[,8:(ncol(tem_rm_na)-1)])
write_tsv(final_out,str_c("./out/4_Rebind_All_Output/",prefix,"_IT_DS_MLM_CPU.Output.final.tsv"))
这一步是为了将最终结果整理输出保存,自动根据项目名称建立结果文件。tsv文件可以直接选择以Excel打开,就是常规表格格式。
查看结果
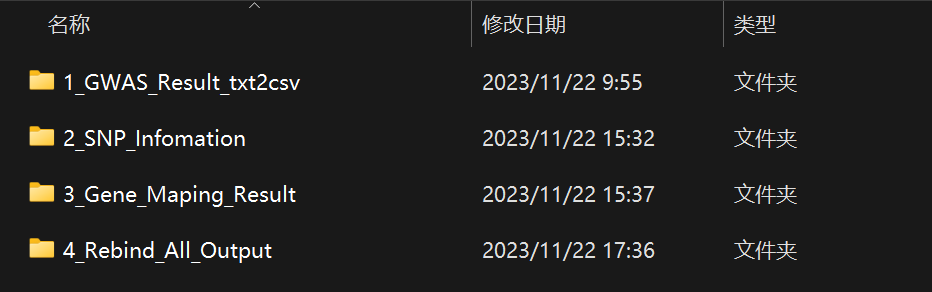
正常情况下,运行完上述流程后,能够在out文件夹发现如上信息。
其中第一个文件夹储存了原始文件转换后的结果,第二个文件夹储存了每个SNP的详细信息,第三个文件夹是显著区域回帖到参考基因组的结果,第四个文件夹内是最终的一个结果文件。
其中最后一个结果文件很重要,包含了所有的显著信息,并对多环境同时共定位到的位点进行标注,可以用于后续研究。
补充:优化思路与方法
在写代码的时候,最开始并没有想到向量化编程的思路,因此在早期版本中采用for循环迭代,速度巨慢。
for (i in 1:nrow(tem)){var_add <- c()for (m in 8:(ncol(tem)-1)){if (tem[i,m] == ""){next}else{var_add <- c(var_add,tem[i,m])}}add_info <- str_c(var_add,sep = "",collapse = " ; ")tem$all[i] <- add_info
}
该流程中最耗时的步骤是对结果进行合并,也就是Step3中将不同年份、地点、类型、模型的显著性关键信息进行整合,合并为一列信息。
在实际计算中,这个数据维度大概是几十万行,每行进行依次迭代的速度很慢,由于计算过程中不需要考虑不同行之间的相互影响,因此考虑改成多线程并行计算,同时在CPU上计算多行数据。
num_cores <- parallel::detectCores() # 设置线程数
cl <- makeCluster(num_cores)
registerDoParallel(cl)
# 原始数据框为tem
n <- nrow(tem)
result <- foreach(i = 1:n, .combine = rbind) %dopar% {var_add <- c()for (m in 8:(ncol(tem)-1)){# 如果出现缺失则跳过if (is.na(tem[i,m])){next}# 如果出现空位则跳过if (tem[i,m] == ""){next} else {# 追加新结果var_add <- c(var_add, tem[i,m])}}add_info <- stringr::str_c(var_add, sep = "", collapse = " ; ")tem$all[i] <- add_inforeturn(tem[i, ])
}
# 关闭并行计算
stopCluster(cl)
registerDoSEQ()
并行计算的速度能有一定提升,理论上64核心处理器的速度会比单纯for循环提高几十倍,但是缺陷也比较明显,这个在计算的过程中每个线程都会复制一份内存空间,导致内存占用量攀升。
最佳的方法是采用R语言向量化编程,使用apply函数直接按行应用函数,这个速度嘎嘎快,而且还节省内存空间。
tem_rm_na$all <- apply(tem_rm_na_info, 1, function(x){x <- na.omit(x) # 删除NA值x <- x[nchar(x) > 3] # 保留字符长度大于3的元素paste(x, collapse = " ; ") # 使用分号作为分隔符连接字符串}
这回看着比较优雅,运行速度也相对提升了一大截。
另外还有一个地方需要进行优化,在不同染色体的分界处需要考虑首尾位置,每条染色体之间是独立的,同一条染色体是按物理位置依次排序,因此确定边界很重要。
以下是原来计算首尾SNP的方法,逻辑是根据当前SNP物理位置与上一行SNP物理位置的大小来比较,如果是结束位置,那么当前SNP减去下一行的值为正值,否则为负值。
if(i == nrow(data)){data$class[i] <- "wei"break}if(a-c<0){data$class[i] <- "shou"next}if(b-a<0){data$class[i] <- "wei"next}
上述算法有个隐藏BUG,当SNP数量多的时候能够正常判断,但是当SNP数量只有几个的时候,有可能会出现某条染色体上最后一个显著的SNP恰好比下一条染色体的第一条SNP位置大,此时算法会将其认为是同一条染色体。
为了解决上述问题,重新修改了判定SNP首尾位置的算法,采用染色体信息直接判断:
# 更新判断SNP首尾位置的方法if (data$chr[i-1] != data$chr[i] &data$chr[i+1] == data$chr[i]){data$class[i] <- "shou"next}else{if (data$chr[i-1] == data$chr[i] &data$chr[i+1] != data$chr[i]){data$class[i] <- "wei"next}else{if (data$chr[i-1] != data$chr[i] &data$chr[i+1] != data$chr[i]){data$class[i] <- "single"next}}}
本文由mdnice多平台发布