🌞欢迎来到AI+生物医学的世界
🌈博客主页:卿云阁💌欢迎关注🎉点赞👍收藏⭐️留言📝
🌟本文由卿云阁原创!
📆首发时间:🌹2024年3月1日🌹
✉️希望可以和大家一起完成进阶之路!
🙏作者水平很有限,如果发现错误,请留言轰炸哦!万分感谢!
人工智能与生物信息学
生物信息学导论及其应用
20世纪后半叶,各种标志性项目开始并达到顶峰对分子生物学领域产生了重大影响。在这些标志性项目的中心是两种生物分子(即蛋白质和核酸),这些项目专注于破译其中携带的信息。
当时被广泛接受的多肽假说指出,每个基因编码一种多肽,将这两种生物分子结合在一起,并建立遗传信息流动的方向。在1955年成功完成第一个测序项目时,通过获得胰岛素蛋白的序列(1,2),Frederick Sanger及其同事强调蛋白质分子是有序氨基酸链。此序列信息进一步有助于确定蛋白质分子在3D空间中的组织。Max Perutz及其同事在破译血红蛋白结构和John Kendrew和在通过X射线晶体学破译肌红蛋白结构方面,一些同事结构生物学领域的里程碑式研究,进一步促进了解决的结构其他蛋白质分子。与我们对蛋白质序列和结构息,在破译核酸分子方面也取得了类似的进展。Avery、McLeod和McCarty在1944年的实验强调了核酸的作用分子作为遗传信息的载体,这后来得到了来自好时和蔡斯在1952年(6)。这些研究之后是Watson的里程碑式发现,克里克和富兰克林在1953年(7)帮助阐明了DNA的双螺旋结构分子。在蛋白质测序工作之后,尽管有一些滞后,Holley和同事成功地对丙氨酸进行了测序,这是第一个核苷酸测序项目1965年转移RNA。后来,Maxam和Gilbert,以及Sanger和同事独立开发了不同的DNA测序技术。这些技术是一个重大的飞跃我们对物种基因构成的理解;然而,它们的一个主要缺点是在处理大量序列数据时缺乏速度。.下一代
测序(NGS)技术基于与Sanger测序技术相似的原理,现在已经通过实施并行测序方法和
已经能够进行各种大规模的测序项目,如物种特异性全基因组测序和个体基因组测序项目。在过去的几十年里,随着技术进步和推测能力的增强,生物信息学对生物科学的影响急剧增加。因此生物信息学已经应用于广泛的领域,如基因组学,蛋白质组学、代谢组学等。
基因组学——生物信息学被用于绘制碎片化输出的关键任务将从测序实验获得的读数与它们各自的来源序列进行比较。单核苷酸多态性(SNPs)和插入和/或缺失的存在基因组片段的(indels)使整个基因组组装的任务特别具有挑战性。这些挑战可以在测序水平上解决(即,通过增加测序深度和/或通过DeepVariant、GATK、VarScan等生物信息学工具,这有助于从测序误差推断出真正的变异。此外通过生物信息学,各种全基因组关联研究能够突出显示可能导致特定表型的基因座/变体。
蛋白质组学——与从测序中确定基因组序列数据类似实验中,生物信息学被用于从输出中推断肽序列数据,通过从头肽测序或通过对照参考数据库搜索质谱(MS)实验的片段数据。结果的功能特性,然后可以通过检查蛋白质标识符的基因本体来执行蛋白质序列条款。在蛋白质标识符不可用的情况下,在线域预测工具可以使用InterProScan来突出内部的潜在功能域蛋白质,从而对蛋白质进行功能表征。除了推断蛋白质的序列和功能注释,生物信息学也被赋予了任务识别潜在的蛋白质-蛋白质相互作用(PPI)网络。各种在线数据库,如
StringDB(21)、BioGRID(22)、IntAct(23)等包含跨不同的物种。在这里,网络中的边缘(两种蛋白质之间的连接)是构建的通过数据管理/实验验证或基于生物信息学的预测
基于多种因素,如蛋白质的共现、共表达、蛋白质同源性等。代谢组学——推断可能导致特定疾病的相关代谢产物表型,包括疾病状况,是来自
代谢组学领域到疾病生物学领域。通过获取途径信息和统计分析,生物信息学已被用于强调代谢产物可能与特定表型有关。生物信息学影响力日益增长的一个重要驱动力也是计算机编程语言的发展。这些语言为导入提供了坚实的平台通过支持广泛数据阵列的读写功能,实现生物数据格式。接下来,一个开源包开发和访问系统能够处理和访问分析这些数据。Python等编程语言的出现使由于它们的高度灵活性、简单的词汇表和开源包管理系统,这些任务更容易完成。生物人工智能
人工智能(AI),一个专注于创建可以执行以模仿人类智慧的方式发挥作用,已经发展成为这一代最有前途的领域。通常,人工智能由一组预先输入的指令组成其执行是以刺激为条件的。人工智能最早的杰出实现之一是深蓝电脑,它在一场比赛中击败了加里·卡斯帕罗夫(当时的国际象棋冠军)1997年的国际象棋比赛。由IBM开发的计算机Deep Blue是基于国际象棋的一套规则。然而,随着对手的一举一动,比赛变得越来越复杂,因为这会带来一系列可能的结果。在这里,计算机的任务是跟踪所有可能的结果,并采取有计划的行动
胜利。尽管深蓝电脑具有象征意义,但它让我们得以一窥人工智能的真正潜力。鉴于生物数据的复杂性,人工智能的应用要求算法不要依赖于一组预定义的规则,而是创建并不断更新可以更好地定义手头的系统。拥有一组从零到最小的预定义的规则(人为干扰)是,推断可以是真正公正的,并反映数据的实际性质。机器学习(ML)是人工智能的一个分支,采用了基于模型的方法其中它从一组“训练”数据中“学习”。根据训练数据的性质,ML算法大致分为两类:有监督学习和无监督学习算法。在监督学习中,训练数据与一组表征数据和算法的任务是建立一组可以定义分配的规则的标签添加到数据中。在无监督学习中,训练数据是未标记的,算法是任务是识别潜在的标签,类似于监督学习,将数据分配给标签。深度学习(DL)是ML的一个分支,是一种先进的人工智能算法,它实现了类似的
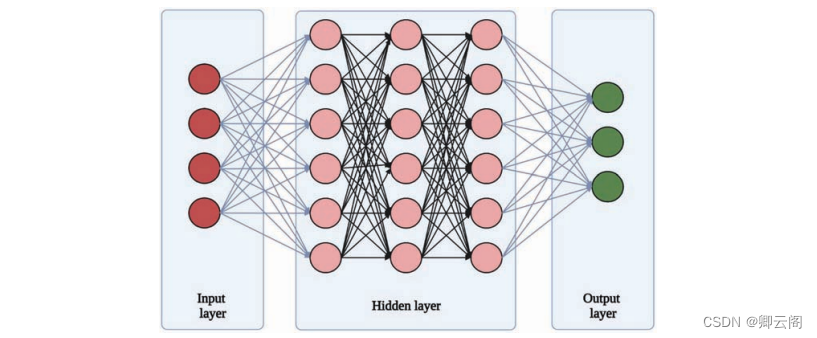
功能与ML相同,并以中高度互联的神经网络为模型人类大脑。典型DL网络的结构由多个互连的“层”组成神经网络通过隐藏将信息从输入层传输到输出层层。隐藏层接收来自源或先前神经的输入层,然后通过激活函数进行处理,并将输出传递给下一个神经层。构成这些层的各个元素被称为感知器. 单个感知器获取加权输入并使用激活函数对其进行处理,并产生二进制输出,然后将其传递到下一个神经层。由于这种复杂性多层结构,DL算法可以识别不同结构之间的非线性关系数据中的变量,从而提高了分类的准确性。
一个典型的神经网络由三层组成,即:输入层、隐藏层和输出层。信息流通过隐藏层从输入层流到输出层,并通过感知器(此处显示为圆形),使用激活函数处理加权输入数据,神经网络方法,卷积神经网络(CNN)算法是基于神经网络方法,并已在处理多维数据中实现,具体而言图像数据。一般来说,人工智能算法已经应用于广泛的领域,如数据挖掘、基因表达谱分析、蛋白质折叠等。
人工智能在解决生物问题中的应用
人工智能算法处理和解释复杂生物数据的能力对解决生物学中许多长期存在的问题产生了重大影响。这些算法的能力处理异构数据使其能够跨各个领域实现,并解决了这些领域的问题。下面列出了其中一些。
蛋白质折叠问题
鉴于蛋白质在细胞生化机制中的核心作用,了解它们结构和序列特征有助于理解各种生物过程。然而预测蛋白质折叠(即根据其序列预测蛋白质的3D结构)是结构生物学领域中具有挑战性的任务。这个问题可以通过同源性来解决建模-使用模板结构构建目标蛋白质的3D结构该序列和靶蛋白序列具有很高的相似性。然而,这种方法在同源模板不可用的场景。AlphaFold(28)是一个ML工具,它使用从蛋白质序列中求解蛋白质结构的神经网络方法,也可以可以在缺乏同源蛋白质结构的情况下使用。蛋白质的结构通过AlphaFold解决的问题可在AlphaFold.ebi.ac.uk/上获得。
人工智能在疾病生物学和个性化医学中的应用
ML在疾病生物学中的应用对医疗保健产生了重大影响。这在生物标志物发现的情况下是特定可见的,其中人工智能算法可以处理多方面的信息,临床数据,可以突出某些疾病特有的特定分子特征条件这些生物标志物可以用来了解疾病的进展。这就处方治疗而言,信息是重要的。除此之外,深度学习算法还通过分析医学图像数据来帮助临床决策,如核成像、超声波、断层扫描等。这些算法已被用于检测癌症的早期发现与转移。
从序列读取中识别基因组变异的问题
全基因组测序实验通常会产生短读序列,这是必需的以映射到参考基因组。如前几节所述这项任务的挑战是区分测序中的错误和真正的基因组变体,因为两者都可能留下相似的签名。DeepVariant采用CNN方法在解决称基因组变异的问题时。DeepVariant使用测序读取数据围绕每一个候选基因组变体,使用图像叠加技术,内部计算每个站点的概率都是真实的变体。HELLO在突出显示时使用了类似的方法遗传变异,但它的算法基于输入测序数据,而不是图像堆积周围读数的。
预测转录因子结合位点
转录因子在基因调控中起着重要的作用。通常,TF与效应基因,以序列特异性的方式,最终导致它们的翻译,最终,表达。转录因子结合启动基因翻译的位点是已知的作为转录因子结合位点(TFBS),这些TFBS内发生的突变可能潜在地切断这些调节相互作用。这些TFBS通常表示为通过各种生物化学测定获得的广义序列每个位置的模糊度。因此,了解TFBS中基因组变异的影响并且定义每个TF的总体绑定模型变得具有挑战性。解决的问题
DeeperBind(30)强调了DNA和RNA结合蛋白的序列特异性最早采用CNN深度学习方法的方法之一。在DeepBind之后,在理解DNA和RNA结合蛋白,如DeepGRN、DeepRAM等。人工智能与化学信息学
化学信息学导论
化学是一个研究元素和物质的科学领域,诞生于130亿左右几年前,当一个豌豆大小的奇点猛烈地扩展到我们今天看到的巨大霸权时作为宇宙。它不仅研究所有不同的物质组合
形成大量的化学结构(原子、分子、化合物等),但也起着从我们使用的牙膏到汽车燃料,在日常生活中发挥着至关重要的作用。这个富人光谱继续扩展到其他方面,如制药、纳米技术,以及作为药物的发现和开发。随着化学的广泛应用需要有效管理的海量数据。这是多学科领域的化学信息学进入画面。系统地组织如此饱和的数据量的下一个垫脚石是挑战这些数据包括结构、小分子、分子式及其性质。这导致延伸到一个新的分支,即今天广为人知的化学信息学。它主要侧重于设计一个系统来管理生成的所有化学数据,以便操纵并将其应用于一系列其他领域以使药物发现更简单、更高效、更高效,有效。
以下部分简要讨论了化学信息学的历史和它的一些应用。可以将化学信息学定义为化学和信息技术的综合,它使用计算技术来管理大量的化学数据,这些数据有助于在化学品和结构元素之间建立联系。它包括这样的概念作为化学数据库,定量构效关系(QSAR),化合物性质或光谱预测等。尽管这些概念已经存在了40多年化学信息学最早由弗兰克·布朗于1998年提出。有必要认识到这是一个独立的分支,因为科学家们正在努力确定这项基于计算机的研究和组织,这使得他们更难将几个学科关联在一起并找到共同的目的。化学信息学不仅允许将数百万种化合物组织成大型所有人都可以访问的数据库,但也有助于解决化学中的复杂问题,如确定化合物的结构与其生物活性之间的关系或反应条件对化学反应性的影响,这需要新的方法从数据中提取知识并对复杂关系进行建模。一些应用化学信息学讨论如下
药物发现
开发具有潜在靶标结合药代动力学的先导化学品的过程,可能被改进以满足美国食品药品监督管理局的安全有效标准被称为药物发现。它包括识别和验证目标以及命中。化学信息学在药物发现中的应用包括化合物选择、虚拟文库生成、虚拟高通量筛选、HTS数据采矿和早期计算机ADMET(吸收、分布、代谢、排泄和毒性)预测。化合物选择主要集中在从
大型游泳池。它通常涉及四个主要目标:
- 能够从其他外部来源挑选和获取丰富多样性的物质。
- 能够从公司复合物库中选择一个子集进行筛选,该子集代表多样性
- 能够选择化学品来创建一个尽可能多的组合库多样性
- 能够选择具有与已知配体相关的新支架的化合物,但是与现有的化合物集合不同
虚拟高通量筛选和实验高通量筛选之间的唯一区别筛选是前者比后者更强调化合物选择。它试图将必须在实验环境中接受测试的申请人数。
在化学信息学中,ML技术通常用于预测生物和可在实际环境中使用的新型化学品的化学特征。团结女神像,182米高的建筑物可以用作对此的具体说明。根据预测氧化会导致棕色结构在大约100年后变绿,类似于自由女神像。几种ML方法,如识别模式,揭示了关系
在化学结构和它们各自的生物活性之间。用于导线识别和优化,采用了多种技术和方法。其中一些包括虚拟筛选、分子数据库、数据挖掘、高通量筛选(HTS)、QSAR、蛋白质
配体模型、基于结构的模型、微阵列分析、性质计算和ADMET属性。ADMET分析的主要目的是了解药物代谢——这一过程人体主要通过化学修饰(也称为生物转化)来去除药物。药物经肝脏代谢后显著减少(细胞色素P450肝脏中的酶通过氧化、水解和羟基化进行代谢)。ADMET这些特性使开发人员能够研究潜在药物的所有特性,以帮助降低临床试验中的潜在风险。它们确定一种化合物是否适合进行临床阶段。开发人员可以了解必要的候选药物的安全性和有效性以供监管部门批准。例如,乙醇很容易从胃肠道吸收,但皮肤吸收不良。HTS是一种实验室自动化检测技术,用于快速筛选大量化学化合物的所需活性。当一个分子与另一个分子连接以产生稳定的复合物时,一种预测分子与蛋白质的优选取向的方法被称为对接. 这种方法包括研究两种大分子之间的相互作用。
计算机辅助综合设计
计算机辅助合成设计(CASD)是化学信息学的另一个领域,需要人工智能技术的应用。化学数据库是用各种类型的机器设计的可理解的化学表示,以便存储其数据,以便以后检索和处理。一个这样的化学格式的例子是简化分子输入线输入规范。这是一种描述连接的线性化学符号格式表和分子的立体化学。化学信息学的另一个方面是结构表示,通常与分子建模、结构搜索、计算机辅助结构阐明等相关。这些涉及将结构作为整体,检查相似性和多样性,并识别关键特征,如结合角度、环和芳香性。结构描述符(拓扑、几何、混合或3D、电子)在考虑的物理、生物和化学性质时也很重要感兴趣的化合物。基于CASD的程序的一个例子是Logic和启发式应用于合成分析(LHASA)。它使用了启发式方法Corey在20世纪60年代末发明,至今仍在使用。它由一种特殊的化学语言组成,称为CHeMistry TRaNslator(CHMTRN),有助于搜索断开连接。这很容易学习语法与英语相似的程序设计语言,因此更便于用户使用对于化学家来说。
化学信息学:从大数据到人工智能
化学信息学主要处理通常存储的化学数据在大型数据库中,并用于推断知识。这些数据可能会变得极其复杂,难以手动解释的多种结构和特性。因此,大数据指通过参考过去和方法和可用于处理它们的设备。
根据《商业分析数据挖掘》一书,存在许多障碍,在分析大数据时要克服的问题,包括数据量和速度生成和更改,生成的数据的多样性,以及数据的准确性是指它是通过有机分布式工艺生产的,不受与收集的数据相同的控制或质量检查。
化学中产生的大数据被称为“BIGCHEM”,包括大型数据库已经通过实验技术,如高通量筛选,平行合成等。为了从这些数据中获得见解,有必要对其进行挖掘以消除在所有噪声中(有时是由于测定过程中的实验错误、缺乏标准化注释生物数据或错误,同时提取数据值、单位、化学类型等)存在于原始数据中。使用各种ML方法来分析这些数据,最终有助于将其可视化。然后可以使用它来预测未来的结果。的这一部分本章将重点讨论人工智能在化学信息学中的作用。
人工智能
人工智能为BIGCHEM提供了帮助。在实际考虑时,许多专家预测人工智能最终将取代人们目前在许多行业所做的工作,导致普遍失业。然而,当人工智能通过最大限度地减少处理化学数据所需的工作量,实现大数据。神经的与大数据相关的网络是广泛使用的人工智能和ML技术的例证化学信息学。这是一种经过时间考验、适应性强的数据驱动分类或预测方法。用于模式识别的数学模型的架构在很大程度上是对其巨大的预测准确性负责。输入层中的许多神经元,即隐藏层或者层,并且输出层将其保持在一起(38,39)。典型结构中神经元之间的每一个环节都有一个权重。在对尚未观察到的实例进行测试之前,权重在训练阶段随着网络学习如何连接输入和输出数据而进行调整。该模型基于人脑的神经网络,与之非常相似,并模仿人类学习的方式。在化学信息学中这些可以用于分析和预测生物活性,毒理学、药理学和物理化学特性,如hERG阻断、水生毒性、药物清除率、pKa、熔点和溶解度。
深度神经网络
深度神经网络(DNN)是深度学习的一个子类型,也是一种非常有用的ML算法
到计算机ADMET分析。它利用了神经网络的几个层,其中每一层取决于前一个。它可以学习不同级别的多个表示级别抽象的(43)。这种强大的算法不仅允许人们根据SMILES化学格式,但也有能力创建新的、更具表现力的表示与已经存在超过28年的传统化学指纹技术相比。支架代表一组中常见的最小芯环子结构的组成化合物。根据用于确定其合成和结构复杂性的各种指标,这些可能会变得极其复杂。过于复杂的命中会导致化学挑战,从而在很大程度上减缓SAR的开发过程。由于对映体和非对映体的体内和体外生物效应不同,解释和分析分子中手性中心的数量变得更加困难。一些用于脚手架分析的算法示例有脚手架树和层次数据库可以使用脚手架树、层次结构和具有线性计算时间依赖性和化学逻辑聚类的类似技术相对更快地进行分析。支架跳跃是另一种在药物发现过程中广泛使用的技术,主要用于化学物质的跳跃通过中心核的修饰来寻找源自已知活性化合物的新的生物活性结构的空间。化学信息学和人工智能在药物发现中的应用、
众所周知,开发新药的过程包括筛选大量有前景的化学文库,然后仔细检查这些文库以产生线索。这些引线然后进行优化,以减轻其负面副作用并增加其有益效果然后通过临床试验进行确认。此程序旨在确保新药快速上市,对患者安全高效。每种药物的市场推广费用通常为1美元亿美元,需要15到20年才能完成。除了昂贵和耗时之外这个过程也异常困难,充满了不确定性。一种出现的铅化学物质。
在早期试验中成为一种可行的药物可能在研究的后期阶段被证明是无效的,导致资源的重大损失。然而,使用人工智能和化学信息学技术可以通过预示将选择哪些化合物来显著降低这种不确定性,以及工作。现代深度学习神经网络技术和强大的人工智能算法(如支持)
向量机已经被证明比传统的ML技术更有前景。一些将简要讨论化学信息学在药物开发和人工智能中的应用。虚拟筛选在药物发现过程的初始阶段起着至关重要的作用通过允许研究人员通过计算或计算机分析分子来预测蛋白质——配体相互作用。这有助于它们消除大量分子,从而减少
实验室实验,也大大加快了这一过程。复合物筛选可以使用各种开源、免费的在线化学信息学工具。一个这样的软件是ZINCPharmer,将药效团搜索筛选技术应用于可供购买的整个ZINC数据库。ZINCPharmer允许用户导入LigandScott和MOE药效团定义及其直接从结构数据库中发现的药效团属性(46)。药效团是分子或药物的特定区域,具有必要的原子和官能团的几何构型以及可以与靶标结合的形状并引发生物反应。定义明确的药效团可以在药物发现过程。由于其速度,基于配体的虚拟筛选(LBVS)仍然是最受欢迎的方法之一以及用于协助药物发现的常用化学信息学应用。它利用配体或与靶标相互作用的小分子的信息来识别新的强效化合物。配体信息可以包括结合亲和力、化学结构、物理化学性质等,ML方法的一些例子是QSAR、相似性搜索和药效团作图。当关于目标结构的信息很少或没有可用信息时,优选LBVS。另一方面,当有足够的靶蛋白结构信息(特别是晶体结构)可用时,使用基于结构的虚拟筛选(SBVS)。它专注于模拟配体与其蛋白质之间的相互作用。分子动力学模拟,对接,同源性建模和药效团建模是所使用的ML方法的一些例子用于这种类型的虚拟放映。
虚拟筛选可以进一步分类为基于药效团的虚拟筛选(PBVS)以及基于对接的虚拟筛查(DBVS)。药效团建模是一种基于结构的方法,是基于“剥离”官能团原有化学组成的概念
根据其主要的物理化学特征将其分类为相对较少的药效团类型(47)。它经常被用来评估分子的概率与蛋白质结合位点结合,并基于相互作用鉴定潜在的结合物。LigandScott是一种用于基于结构和配体的药效团建模的通用软件,它涉及新的高性能比对算法,具有卓越的预测质量前所未有的筛选速度(47)。它允许的自动构建和可视化来自大分子/配体复合物的结构数据的3D药效团。氢键在LigandScott算法的化学中,供体和受体由定向矢量表示特性,以及正和负可电离区域,亲脂性区域的球体,以及氢键供体和受体。此外,LigandScott模型结合了空间关于任何潜在配体都无法获得的区域的数据,增加了选择性并反映潜在的空间限制(48)。生成的药效团模型自动更新为包括被排除在外的空间禁区。相反,基于对接的虚拟筛选侧重于分子的计算拟合使用先进的算法进入受体活性位点,然后对这些分子来识别潜在的线索。通常,该方法从的3D结构开始目标蛋白,然后使用将数据库中的化学物质对接到目标蛋白中对接软件(47)。以最佳方式与受体相互作用的配体是
一旦对对接结果进行了评分,就被选择用于进一步优化。之后,配体通常进行各种结合测定。一个流行的对接软件是AutoDock,它允许预测小分子(底物或引线)与已知受体的结合
3D结构。蛋白质-配体对接软件的另一个例子是GOLD(27)-Genetic配体对接的优化——基于遗传算法对接柔性配体进入蛋白质结合位点。在DBVS中,靶蛋白的3D结构至关重要,必须通过实验了解(X射线晶体学技术、NRM研究等)或使用计算技术(人类学建模)(49)。化合物数据库源(例如,ZINC)通常与
可从化学品供应商处获得的化合物集合。硅过滤器(基于Lipinski规则五)用于减少口服生物利用度低的化合物的数量。进一步检查分子,可以使用过滤器来消除化学稳定性、反应性或毒性较差的物质。之后,根据配体的结合亲和力对配体进行评级(假设为独立项的总和)。在此基础上,根据相对各种配体的结合亲和力,而配体是根据溶液的质量排列的(49). 下一步是对化合物进行后处理阶段的分析,以确保它们与靶蛋白的相互作用和构象是合适的。这些化合物是然后送去化验。桥接生物信息学和化学信息学方法
生物信息学方法
人类基因组计划(HGP)可以被视为生物学领域,特别是对生物信息学的发展起到了重要的推动作用领域测序实验的大量数据积累提出了两个重要问题挑战:数据存储和分析。生物信息学已经开始变得越来越重要在20世纪后半叶,它成为有助于克服的关键领域之一
这两个挑战。今天,提交和检索各种生物数据的任务专用数据库为实验提供了便利。这些数据库中的大多数都是免费访问的并由专家维护和策划。除此之外,一些数据库如Ensembl(其余。ensembl.org/)、UniProt(www.UniProt.org/help/proprogrammatic_access)等也允许程序通过REST-API端点对其数据进行自动访问。通过启用对可用数据的开放访问以及托管来自世界不同地区的数据,这些数据库处于最前沿解决生物问题,特别是需要立即采取行动的问题。举例来说,自从严重急性呼吸系统综合征冠状病毒2型爆发的早期阶段以来,对样本进行测序就变得势在必行并公开这些序列进行分析,以更好地表征病毒并控制其传播。在这种情况下,个人贡献者共享的数据在数据库方面,特别是GISAID(50(GISAID.org/),一直是解决流行病在本章中,我们将讨论各种此类数据库。然而,除了在生物数据集的存储和访问中发挥关键作用外,生物信息学在解释这些数据集中包含的信号方面发挥了更重要的作用生物分子。这种基于生物信息学的方法已经在广泛的字段/域。这里将详细讨论其中的一些方法/应用程序。
人类健康
人类基因组计划(51)估计耗资约30亿美元。然而,由于预付款在测序技术中,对整个人类基因组进行测序的总体成本已经降低在过去20年中显著增加(图1.3)。测序价格的下降幅度也是超过了假设的摩尔定律(52)分布,该分布表现出计算能力每年翻一番。基于生物信息学的基因组组装管道在使得能够如此容易地重建物种的整个基因组方面提供了显著的促进。到例如,CANU(53)是一种基因组组装工具,使用PacBio或Oxford的测序读数
以纳米孔为起点生成全基因组序列。作为附加封装,HiCANU依赖于CANU的内部算法,但使用从PacBio生成基因组组装。NECAT(54)是最近引入的基因组组装工具使用从重建整体的纳米孔测序技术中获得的长读数基因组序列以降低错误率。BUSCO(55)是基因组质量评估工具通过检查组装基因组的“完整性”来组装。今天,基因组测序已经在商业市场上发展了它的利基市场。各种风险投资已经开发出复杂的管道,可以执行敏锐的全基因组测序来自世界各地的参与者获得一个可行的价格范围,并以这样的方式记录结果没有生命科学背景的人也可以理解它们。这些结果包括推断祖先、预测各种特征,以及重要的易感性
对各种疾病。诊断遗传疾病和易患疾病的能力最近通过能够监测基因组及其后果而显著增长
下游产品,如RNA和蛋白质。与基因组测序平行,这些技术最近,参与基因组编辑的研究人员在其靶向特定基因组位置的精确性。生物信息学在这里得到了广泛的应用
以将精确的基因组区域识别为感兴趣的靶标。具体而言CRISPR系统及其应用CRISPR-Cas9,一种主要参与细菌防御的免疫蛋白病毒,彻底改变了基因组工程。个性化医学领域受益匪浅这主要归功于基因组测序和编辑方面的这些进步。为患者开处方通过基因组编辑进行治疗和应对疾病将重塑医学方式治疗。如前所述,生物信息学一直处于记录和存储的前沿
基因组序列。1000个基因组项目(56)(www.internationalgenome.org/)是一个数据库
致力于对通过各种人类基因组获得的序列和变体信息进行编目来自不同地理区域和人群的测序实验。这种群体特异性变体注释有助于理解潜在的有益和有害等位基因(图1.3)
桥接生物信息学和化学信息学公开的方法数据库

本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.hqwc.cn/news/503570.html
如若内容造成侵权/违法违规/事实不符,请联系编程知识网进行投诉反馈email:809451989@qq.com,一经查实,立即删除!相关文章
使用 Docker 部署 Fiora 在线聊天室平台
一、Fiora 介绍
Fiora 简介 Fiora 是一款开源免费的在线聊天系统。 GitHub:https://github.com/yinxin630/fiora Fiora 功能
注册账号并登录,可以长久保存你的数据加入现有群组或者创建自己的群组,来和大家交流和任意人私聊,并添…

day03-Vue-Element
一、Ajax
1 Ajax 介绍
1.1 Ajax 概述 概念:Asynchronous JavaScript And XML,异步 的 JavaScript 和 XML。 作用: 数据交换:通过 Ajax 可以给服务器发送请求,并获取服务器响应的数据。异步交互:可以在 不…
SpringBoot 整合WebService
文章目录 WebService1.简单介绍WebService1.1. 类型1.2. 架构1.3. 主要特点1.4. 使用场景1.5. Web服务标准和技术 2.案例-WebServiceDemo2.1.引入配置文件2.2.创建接口2.3.创建接口实现类2.4.创建WebService配置类2.5.测试 WebService Web服务(Web Services…
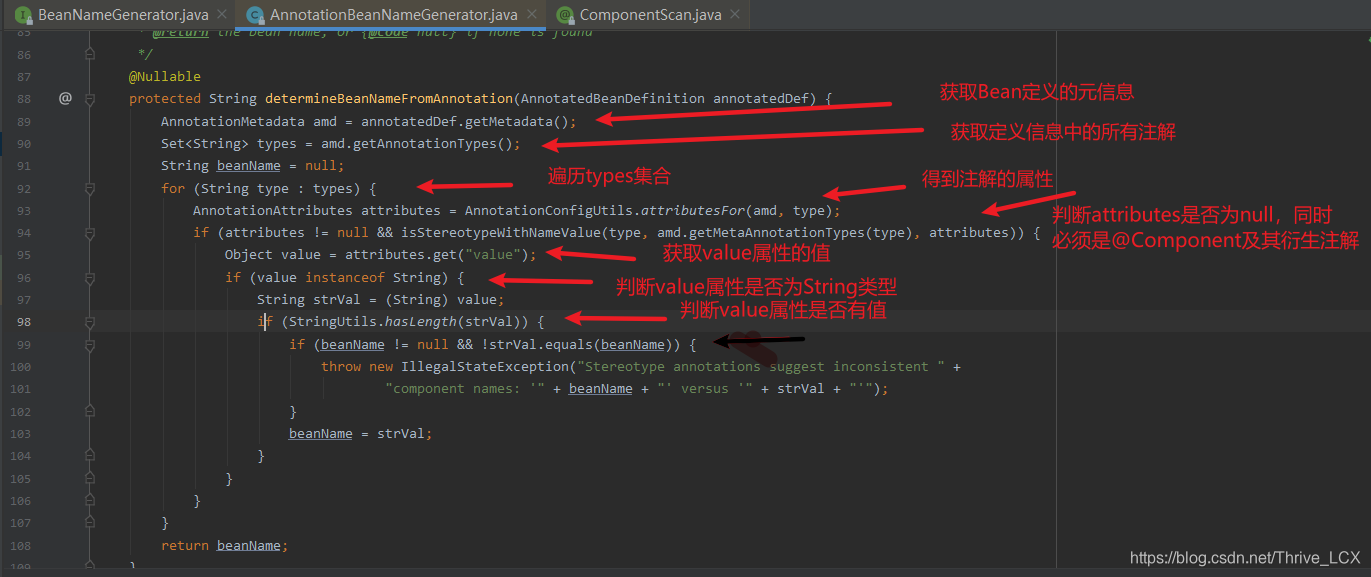
自定义BeanNameGenerator生成规则
通过点进ComponentScan注解进入源码可以看到 追随BeanNameGenerator进入源码可以看到该类是个借口且只有一个方法 点击上面黑色箭头出现两个实现方法
点击第一个方法 进入determineBeanNameFromAnnotation方法中
通过上诉自定义一个生成beanName方法
先创建一个CustomeBeanN…
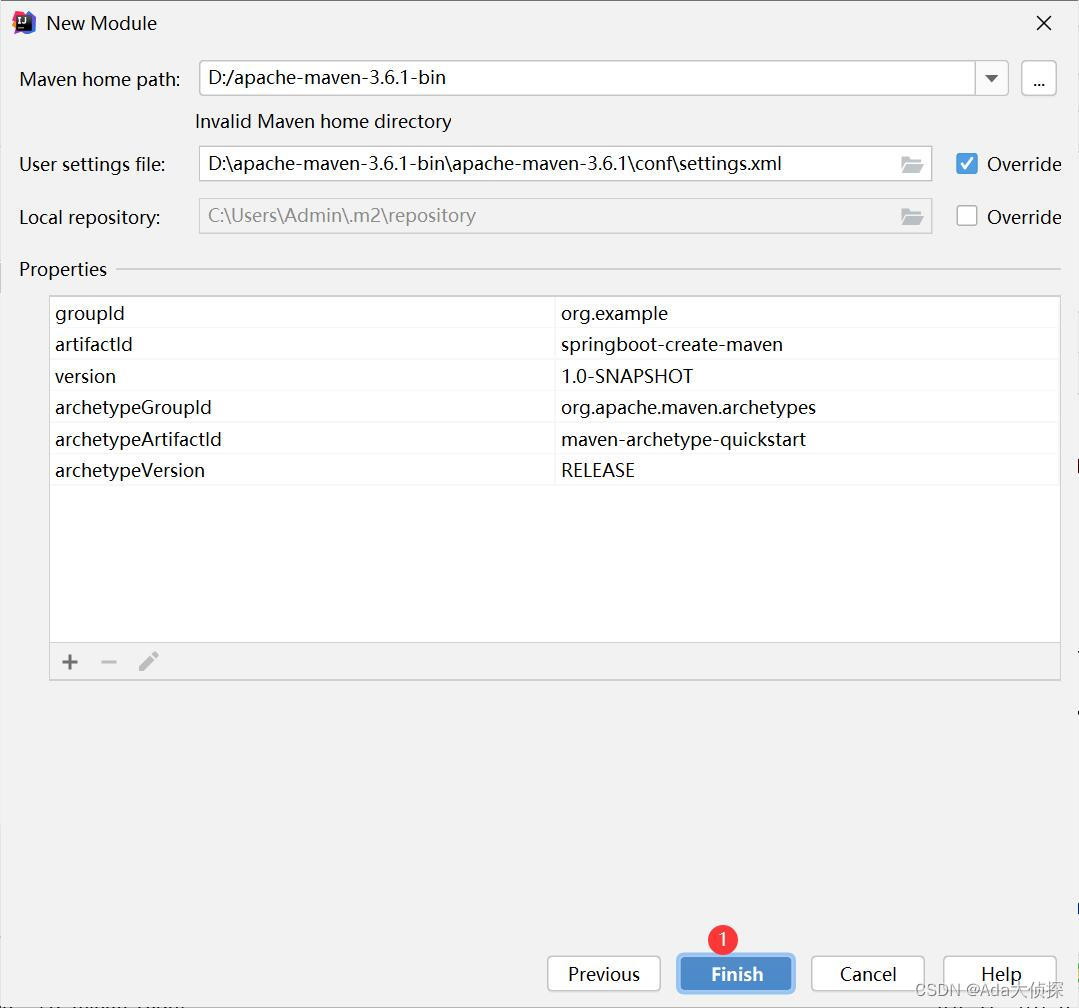
idea使用maven创建springboot项目
按照图片中的流程来,就可以创建springboot项目,我这个主要是想做一个JavaWeb项目
有用的话,点个小赞赞再走呀~

b站小土堆pytorch学习记录——P7-P8 Tensorboard的使用
文章目录 一、前置知识1.Tensorboard是什么2.SummaryWriter3.add_scalar()4.add_image() 二、代码1.一次函数2.蚂蚁和蜜蜂图片 一、前置知识
1.Tensorboard是什么
TensorBoard 是 TensorFlow 的可视化工具,它允许开发者可视化模型的图(graph࿰…
动态规划|【路径问题】|931.下降路径最小和
目录
题目
题目解析
思路
1.状态表示
2.状态转移方程
3.初始化
4.填表顺序
5.返回值
代码 题目
931. 下降路径最小和
给你一个 n x n 的 方形 整数数组 matrix ,请你找出并返回通过 matrix 的下降路径 的 最小和 。
下降路径 可以从第一行中的任何元素开…

力扣SQL50 大的国家 查询
Problem: 595. 大的国家 Code
select name,population,area
from World
where area > 3000000 or population > 25000000;
Apache Echarts介绍与入门
介绍
Apache ECharts 是一款基于 Javascript 的数据可视化图表库,提供直观,生动,可交互,可个性化定制的数据可视化图表。 官网地址:https://echarts.apache.org/zh/index.html
入门案例
Apache Echarts官方提供的快…
三天学会阿里分布式事务框架Seata-seata事务分组介绍
锋哥原创的分布式事务框架Seata视频教程:
实战阿里分布式事务框架Seata视频教程(无废话,通俗易懂版)_哔哩哔哩_bilibili实战阿里分布式事务框架Seata视频教程(无废话,通俗易懂版)共计10条视频&…
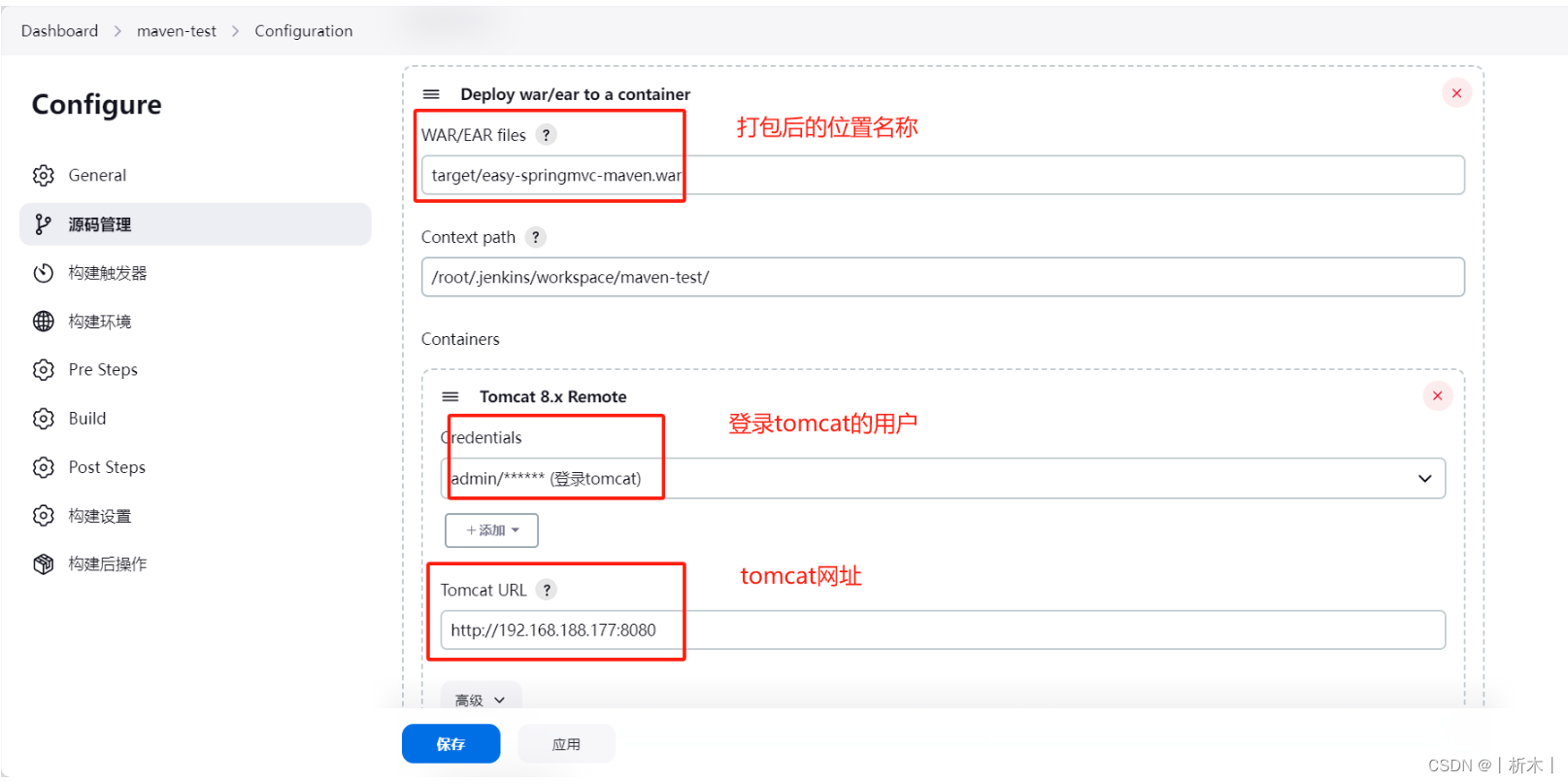
linux系统Jenkins工具添加自由项目和maven项目
Jenkins添加自由项目 添加自由项目操作流程代码远程代码邮件标题邮件正文 添加maven项目准备环境操作流程 添加自由项目
gitlab配置基本代码页面等,拉取代码,打包,发布操作流程 代码
远程代码
echo
ssh root192.168.188.177 "tar cz…