Analyses of a chromosome-scale genome assembly reveal the origin and evolution of cultivated chrysanthemum
分析染色体级别的基因组装配揭示了栽培菊花的起源和进化
六倍体植物基因组的文献,各位同仁还有什么有特色的基因组评论区留言~

摘要
菊花(Chrysanthemum morifolium Ramat.)是一种具有重要经济、文化和象征价值的全球重要观赏植物。然而,由于其复杂的遗传背景,对菊花的研究面临着挑战。在这里,我们报道了一个几乎完整的 菊花 组装和注释,包括27个拟染色体(8.15 Gb;scaffold N50为303.69 Mb)。比较和进化分析揭示了大约600万年前(Mya)菊花属种共享的一个全基因组三倍体(WGT)事件,以及大约300万年前 菊花 可能的特异系列多倍体化。多级证据表明 菊花 很可能是一个片段性异源多倍体。此外,基因组学和转录组学方法的结合展示了 菊花 基因组可以用来识别底层关键观赏性状的基因。CmCCD4a 的系统发育分析追溯了栽培菊花花色育种的历史。这项研究生成的基因组资源有助于加速菊花的遗传改良。
引言
栽培菊花(Chrysanthemum morifolium Ramat.,中文名“菊花”)是一种多年生草本观赏植物,属于菊科(也称复合花科),这是双子叶植物中最大的一个家族,拥有超过1600个属和25000种物种。除了其杰出的美丽外,栽培菊花在作为菊科的代表时,还在开花植物的进化位置上占据关键地位。菊花起源于中国,已有超过三千年的栽培历史。从公元8世纪到17世纪,数个菊花品种相继被引入日本和欧洲,进一步扩展了它们作为观赏用途的育种。目前,由于其花型和颜色的显著多样性,菊花是全球最经济重要的花卉植物之一,占据了花卉产业生产的大部分比例,用途包括切花、园艺和盆栽。此外,由于其营养和生物活性成分,菊花在医药、食品和饮料工业中也有广泛应用。
菊花属拥有超过41种不同染色体倍性的物种,从2n=2x到10x不等,且在该属中天然杂交普遍存在。栽培菊花是一种自交不亲和的复杂六倍体,通常显示出54的染色体数。然而,菊花是否应被遗传分类为自体多倍体或异源多倍体,长期以来一直存在争议。大量研究表明,几种野生菊花物种可能对当前复杂的杂交菊花品种有贡献。因此,经过长时间的驯化和人工选择,栽培菊花现在显示出比菊花属的其他物种更高的观赏特性多样性。然而,菊花属的进化历史尚不清楚,栽培菊花的实际祖先仍然是一个谜。
可用的高质量参考基因组序列是阐明物种起源和进化历史以及其表型多样性的遗传基础的关键,这进一步可以加速未来的育种过程。尽管栽培菊花在经济上和进化上都非常重要,但由于其多倍体性、高重复性、高杂合性和大尺寸,其基因组尚未被解密。最近,报道了两个脚手架级别的菊花基因组,即C. nankingense和C. seticuspe,以及三个染色体级别的基因组,即C. seticuspe,C. makinoi和C. lavandulifolium。这些宝贵资源允许我们解析菊花属的进化历史。然而,这些基因组都来自二倍体野生物种,因此缺乏菊花品种的关键遗传信息,使它们不太适合阐明六倍体栽培菊花表型变异的复杂遗传基础。几项转录组研究使我们初步分离出了一些参与调控菊花观赏性状的同源基因,但由于缺乏栽培菊花的参考基因组序列,通过正向遗传学方法揭示这些性状的新决定因素仍然具有挑战性。
在这里,我们使用来自盆栽品种“钟山紫桂”的单倍体系列(n=3x=27)呈现了一个菊花的染色体级别基因组组装。我们推断最近的全基因组三倍化(WGT)和较小规模的复制有助于扩展与菊花花发育相关的已知基因,并识别了几个与花瓣形状相关的候选基因。此外,我们基于CCD4a基因的系统发育分析追溯了栽培菊花的花色进化历史。菊花参考基因组提供了对菊花属进化的见解,并对未来的菊花遗传和分子生物学研究具有价值。
结果
基因组测序、组装和注释
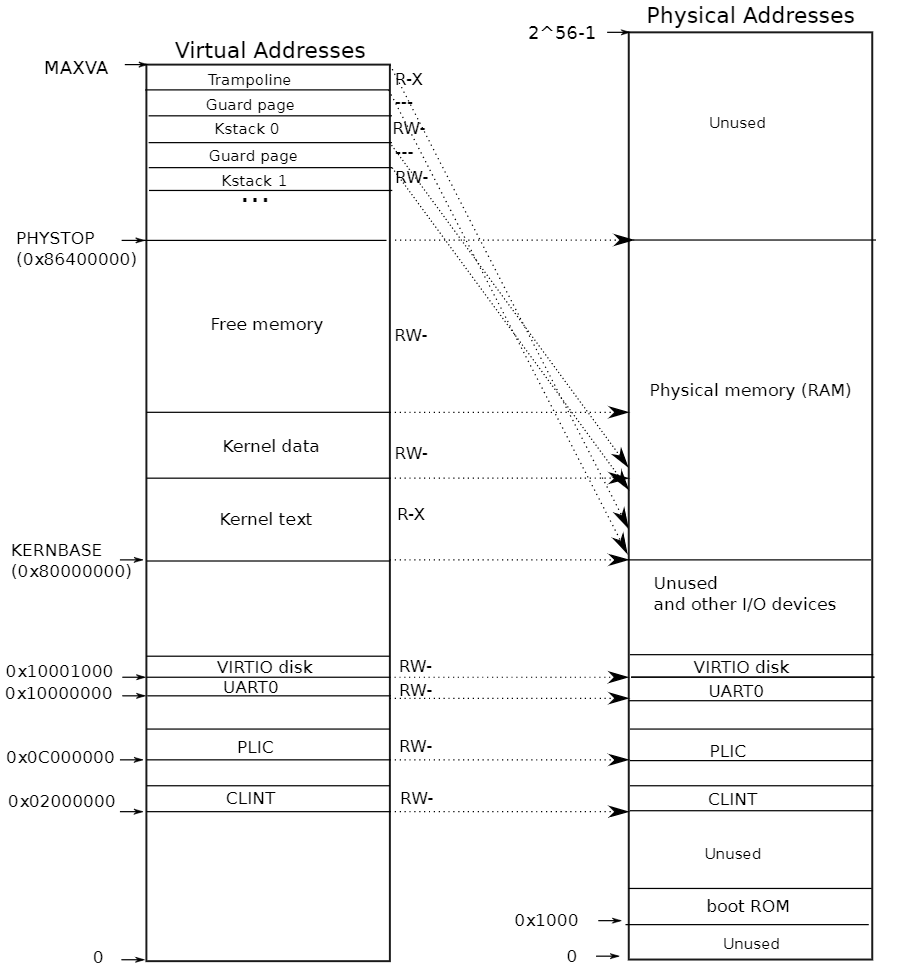
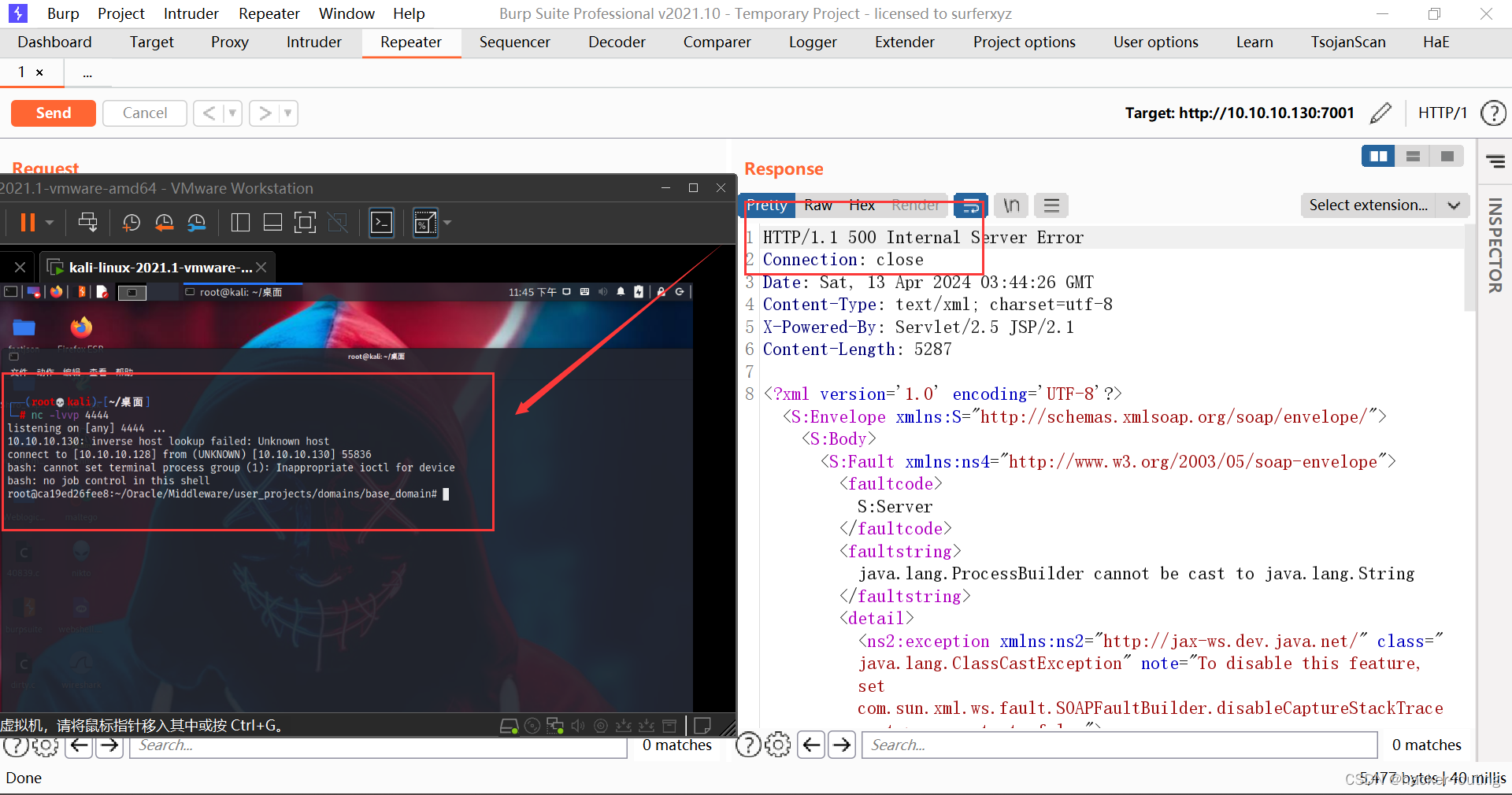
通过使用1070.20 Gb的清洁短读取(Supplementary Fig. 2a–d 和 Supplementary Tables 1 和 2)进行K-mer分析,估计单倍体系列(Supplementary Fig. 1b, d)的基因组大小约为8.47~8.88 Gb,这比流式细胞术估计的大小(9.02 Gb;Supplementary Fig. 2e)略小。基因组调查还揭示了菊花中高重复内容的比例(72.48% ~ 88.85%)(Supplementary Table 1)。我们最初使用Falcon校正并组装了1022.3 Gb(120.70×)的PacBio连续长读取(CLR)成为contig序列,并使用Quiver进行了打磨。然后,通过与907.5 Gb(107.20×)来自10X Genomics库的基因组序列整合并使用短读取进行打磨,对一致性序列进行了支架。接下来,我们使用AllHiC算法根据1002.9 Gb(118.50×)的Hi-C数据(Supplementary Table 2 和 Supplementary Note 1)改进了菊花基因组组装。这允许将原始组装序列的96.46%锚定到长度范围从214.51到343.67 Mb的27个拟染色体上(Fig. 1a, Table 1 和 Supplementary Tables 3 和 4)。菊花最终染色体级基因组组装的contig和scaffold N50大小分别为1.87 Mb和303.69 Mb(Table 1)。Hi-C相互作用热图清楚地展示了这27个拟染色体可以被聚类到九个同源组中(Fig. 1b)。同源组内的三个拟染色体在大小、基因数量和重复元素内容方面高度相似(Supplementary Table 4)。每对同源基因对的保守共线性关系和Ka/Ks比率的图表清楚地显示了高度的保守共线性,且任意两条染色体之间的整体Ka/Ks比率没有显著差异(Fig. 1a)。
一个展示单倍体菊花 cv. ‘钟山紫桂’基因组特征的环形图。从外到内的环形轨迹表示:a 拟分子;b 基因密度;c 长终端重复(LTR)转座子密度;d Copia转座子密度;e Gypsy转座子密度;f DNA转座子密度;g GC含量;h 在同源染色体组内,traid1与traid2或traid3之间识别的同源基因对的Ka/Ks;i 在同源染色体组内,traid2与traid1或traid3之间识别的同源基因对的Ka/Ks;j 在同源染色体组内,traid3与traid1或traid2之间识别的同源基因对的Ka/Ks;k 不同拟染色体之间的同源基因。橙色线条表示traid1与traid2和traid3之间的同源块,绿色线条表示traid2与traid3之间的同源块。b Hi-C热图显示染色体相互作用。每个同源组包含三个拟分子,不同同源组之间的连接很少,表明高质量的染色体级支架。c 菊花(Chr1~27)与二倍体C. nankingense(Cna1~9)之间的基因组比较。来源数据提供为源数据文件。
为评估组装基因组的质量,我们将从美国国家生物技术信息中心(NCBI)数据库下载的105,996个菊花表达序列标签(ESTs)与组装的基因组进行了比对,发现96.43%的ESTs能被映射(Supplementary Table 5)。BUSCO和核心真核基因映射方法(CEGMA)分析发现,分别有97.70%和98.39%的总真核保守基因在菊花的染色体级基因组中被识别(Supplementary Table 6),这与C. seticuspe基因组的结果相当,但高于其他已发布的菊科物种基因组的百分比(Supplementary Data 1)。长末端重复(LTRs)的注释揭示了一个LTR组装指数(LAI)得分为27.99,达到参考基因组的黄金标准,与四倍体紫花苜蓿(22.30)和异六倍体草属植物(22.5)的得分相当。使用Illumina测序数据评估的高映射率98.81%和高基础准确度估计(每1 Mb有57个纯合单核苷酸多态性(SNPs)(0.0057%))也支持了最终组装的高保真度(Supplementary Table 7)。提取并针对组装的基因组映射了最长的10×(约80 Gb)PacBio长读取,范围从36.32到156.36 kb,大多数(82.24%)的这些读取可以唯一映射到只有一个染色体上,且对齐长度超过80%,表明大部分染色体被正确相位。此外,82.15%的转录组能被映射回最终组装(Supplementary Table 8),进一步支持高水平的基因组完整性。综上所述,以上结果表明菊花基因组具有高度的连续性和完整性。
此外,使用类似的杂交组装策略生成了二倍体C. nankingense(2n = 2x = 18)的染色体级组装,这是栽培菊花的潜在祖先,用于比较研究(Supplementary Table 9 和 Supplementary Note 2)。基因组组装将2.87 Gb的contigs锚定到9个拟染色体上,代表了估计的3.09 Gb C. nankingense基因组的93.16%(Supplementary Table 10)。高contig N50长度为5.98 Mb和scaffold N50长度为353.78 Mb表明与先前发布的脚手架级C. nankingense基因组相比,是一个优越的基因组组装。正如预期的那样,互基因组比较分析揭示了菊花和C. nankingense之间明显的3对1的共线性关系。只识别了少数倒置或易位区域(Fig. 1c)。
基于de novo和同源性预测以及转录组数据(Supplementary Note 3),我们在菊花基因组中预测了总共138,749个蛋白编码基因,这比为其他菊科植物注释的基因数量多得多,范围从环球朝鲜蓟的28,310个基因到C. seticuspe的74,259个。总基因区域、编码序列(CDSs)、外显子序列和内含子序列的平均长度分别为3912、1090、242和806 bp(Supplementary Data 2)。平均每个预测基因包含4.50个外显子,整个基因组的基因密度大约为每58.74 kb一个基因,其中134,450个基因(96.90%)位于染色体锚定的contigs上。如预期,基因分布不均,朝向染色体臂的末端更为丰富(Fig. 1a)。大约99.3%的预测蛋白编码基因可以通过在NR(90.97%)、Swiss-Prot(75.58%)、GO(90.78%)、KEGG(70.28%)、InterPro(98.75%)和Pfam(72.60%)数据库的搜索中进行功能注释(Supplementary Table 11)。此外,我们识别了50,848个非编码RNA(ncRNA)基因,编码2868个核糖体RNA(rRNAs)、4102个转移RNA(tRNAs)、2280个微RNA(miRNAs)和41,598个小核RNA(snRNAs)(Supplementary Table 12)。使用来自花蕾和九种器官(包括根、茎、射击叶和从辐射小花和盘状小花中解剖出的花器官)的RNA-seq数据,103,287(74.44%)的已识别基因在至少一种组织中表达,57,869(41.71%)在所有分析的器官中表达(Supplementary Table 13)。
比较基因组学和进化分析
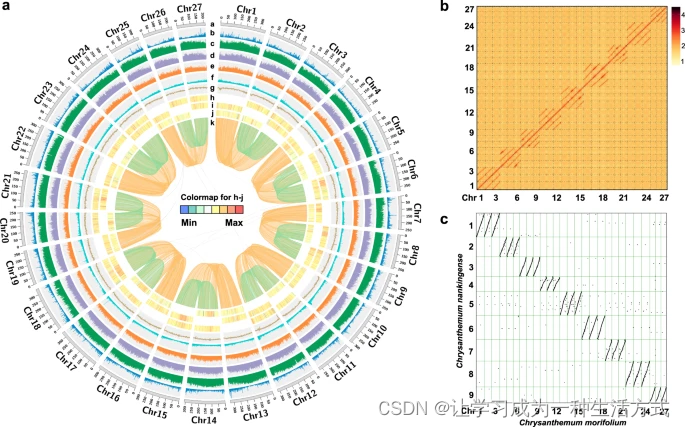
我们推断了栽培菊花与其他14种植物物种(Supplementary Note 4)的系统发育位置和分化时间,包括7种唇形花序I(C. nankingense, C. seticuspe, 向日葵, 生菜, 环球朝鲜蓟, 黄花蒿 和 胡萝卜),2种唇形花序II(番茄 和 咖啡),1种蔷薇目(葡萄),1种毛茛目(耧斗菜),2种单子叶植物(水稻 和 玉米)以及1种基部被子植物(珊瑚树)。我们构建了一个包含94,552个基因的共识集,以代表菊花的单倍体基因组。总共507,449个基因被聚类到48,644个同源基因家族(orthogroups)中,包括6193个由所有15个物种共享的基因家族(Supplementary Fig. 3 和 Supplementary Data 3)。通过比较七个菊科物种,我们识别了所有成员共享的10,234个基因家族和3543个家族,这些家族包含9638个基因,看起来是菊花独有的(Supplementary Fig. 4)。此外,基因家族进化分析揭示了1684个基因家族在菊花中可能发生了扩展,而1926个基因家族显示出收缩(Fig. 2a)。功能富集分析表明,这些扩展的基因家族主要涉及到萜类生物合成过程、生长素代谢过程、激素水平的调节、花青素生物合成过程的调节以及通过同源重组修复双链断裂的调节(Supplementary Fig. 5 和 Supplementary Data 4)。
a 系统发育树展示了栽培菊花与其他14种开花植物的进化关系。基因家族数量的扩张(绿色)和收缩(红色)显示在右侧。一些节点上的红色圆圈代表化石校准点,这些点从TIMETREE网站(TimeTree :: The Timescale of Life)获得。预测的分化时间(百万年前,Mya)以蓝色标记在其他中间节点上,估计是通过最大似然(PAML),括号中的数字是不同类群之间分化时间的95%置信区间。全基因组复制(WGD)和全基因组三倍体化(WGT)事件用彩色点标记。b 菊花和其他五种已测序菊科物种内同源旁系基因的Ks分布。多倍体化事件参照峰值。c 菊花与C. nankingense、A. annua、H. annuus、C. cardunculus之间的重复序列比较。d 不同菊科物种中完整LTRs的插入时间分布。源数据提供为一个源数据文件。
正如预期的那样,使用491个单拷贝基因家族构建的系统发树(图2a)与先前的研究一致。分子定年表明,菊科(Asteraceae)与胡萝卜(Apiaceae)大约在92.4百万年前分化。在菊苣族(Anthemideae)内,栽培菊花从C. nankingense和C. seticuspe的最近共同祖先分化出来约在3.7百万年前,这是在大约6.5百万年前黄花蒿(Artemisia annua)分化之后。菊苣族物种与向日葵(Heliantheae,大约在34.8百万年前分化)的关系比与生菜和环球朝鲜蓟(Cichorieae,大约在39.3百万年前分化)更为密切。
众所周知,全基因组复制(WGD)或全基因组三倍体化(WGT)事件对于塑造植物基因组的进化和物种形成具有深远影响。我们首先估计了菊花、C. nankingense和C. seticuspe、向日葵和环球朝鲜蓟内的共线旁系基因对的同义替换率(Ks)值,以识别WGD/WGT事件(图2b)。这些结果与先前的报告一致,表明菊花具有复杂的古多倍体历史。除了所有核心真双子叶植物共享的古老WGT-γ事件(大约122~164百万年前),唇形花序II共享的WGT-1(大约57百万年前)和向日葵中的WGD-2(大约38百万年前),我们识别了一个最近的,三个菊花属物种共有的特异系列WGD事件,大约发生在6百万年前,显示出大约0.1的典型Ks峰值,与先前的研究一致。此外,基因组点图分析清楚地展示了菊花单倍体拟染色体内的3:1共线关系和菊花与环球朝鲜蓟之间的3:1共线关系,后者只经历了菊科共享的WGT-1事件。在C. nankingense内部以及C. nankingense与环球朝鲜蓟之间检测到了类似的关系。这些结果提供了额外的证据,表明菊花属共同祖先中推断的最近多倍体化事件更可能是WGT事件而非WGD事件(此处称为WGT-2)。我们还计算了每个九个同源组内旁系基因对的Ks值,并在大约0.05(大约3百万年前)发现了另一个典型峰值,表明除了三个菊花属物种共享的特异系列WGT-2事件外,菊花经历了一个非常近期的多倍体化事件。这可能是观察到的菊花基因组具有高度保守的三倍体结构而没有显著染色体重排的一个潜在原因。
基因复制被认为是进化的主要动力。在菊花基因组中,136,137个重复基因被划分为五个类别,即全基因组复制(WGD, 61.5%)、分散复制(DSD, 43.1%)、转位复制(TRD, 17.0%)、近端复制(PD, 6.9%)和串联复制(TD)。发现TRD和PD基因对具有更高的Ka/Ks比率,表明转位和近端复制是一个持续进行的过程,并且通过这两种方式生成的重复体受到了更松弛的选择压力,而来自所有五种方式的复制显示出类似的Ks值。值得注意的是,通过所有五种方式的复制与不同的GO项富集相关。例如,‘WGD基因’显著富集于各种发育过程、转录调节和信号转导的GO项;PD基因富集的类别涉及到花粉识别和倍半萜生物合成过程,而TD基因被分配到单萜生物合成和萜类代谢过程以及应激反应。萜烯合酶(TPSs)是绿色植物中萜类化合物生物合成的关键酶。我们发现菊苣族物种的基因组比其他菊科植物含有更多的TPSs,特别是对TPS-a和TPS-b亚家族,这些主要涉及倍半萜和单萜的合成。识别了几个串联重复的TPS-a和TPS-b基因。特别是,13个TPS-a副本和10个TPS-b副本分别密集分布在27号和19号染色体上。
通过ab initio和同源性方法的结合识别了重复序列。总共83.38%的组装序列被注释为重复元素(TEs),包括79.59%的逆转录转座子、8.63%的DNA转座子和0.92%的简单重复和卫星。据我们所知,菊花中识别的TEs比例是已测序菊科物种中最高的,从环球朝鲜蓟的58.4%到向日葵的74.7%不等。LTR逆转录转座子被发现构成了菊花基因组的72.96%,其中Copia(40.40%)和Gypsy(24.79%)亚家族占最大百分比。Copia元素密度从染色体的末端向中心增加,而Gypsy元素在染色体上的分布相对均匀。值得注意的是,Copia和Gypsy在菊科中的比例存在较大差异。与菊花(1.63)、C. nankingense(1.74)、C. seticuspe(1.28)、C. makinoi(3.66)和C. lavandulifolium(1.3)基因组中Copia元素更为丰富的情况相反,黄花蒿(0.86)、向日葵(0.26)和新发布的Mikania micrantha(0.37)基因组含有更多的Gypsy元素。这些发现表明,LTR/Copia元素的扩增是菊花基因组膨胀的主要驱动力。Copia和Gypsy元素的这些不同行为的机制尚待阐明。我们发现在这三个物种分化后,菊花、C. nankingense和向日葵中的LTRs发生了非常近期的爆发(<0.5百万年前),这比在黄花蒿(大约1百万年前)和环球朝鲜蓟(大约2百万年前)中识别的LTR爆发略晚。此外,菊花中Copia和Gypsy逆转录转座子插入的爆发发生在接近的时间,且在Copia/Gypsy爆发的预测时间上,各染色体之间没有显著差异。总体而言,这些结果表明菊花基因组的特点是活跃的转座,并且TE插入的积累是栽培菊花基因组扩展的主要原因。
栽培菊花的起源
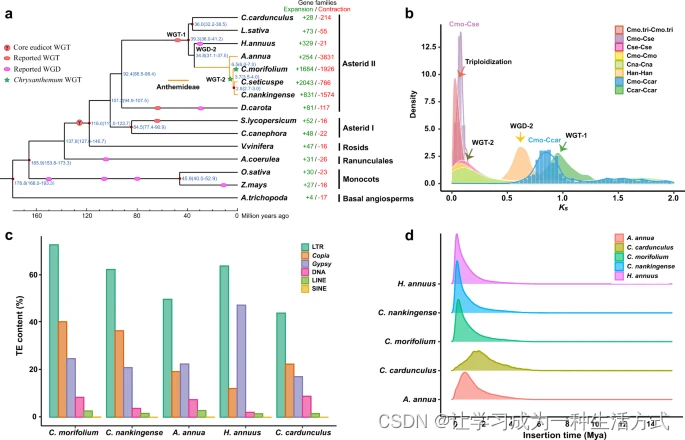
尽管栽培菊花的起源引起了广泛关注,但贡献给栽培菊花的野生祖先仍不确定。旨在识别这些祖先物种的先前系统发育学研究,通常基于传统的形态学和细胞学分类或有限数量的分子标记,产生了不一致的结果。通过利用菊花基因组,我们重新测序了12种被认为是栽培菊花最可能的祖先物种的中国野生菊花物种,每个存取平均覆盖深度约为8.5倍(Supplementary Fig. 13, Supplementary Data 5)。用11,755个SNP生成的系统发育树(图3a)清楚地显示,具有白色辐射小花的C. rhombifolium和来自南京的四倍体C. indicum可能与栽培菊花的关系更为密切,这与发现C. rhombifolium和C. indicum(南京)与菊花共享最多特异片段的结果一致(图3b)。此外,身份得分(IS)分析清楚地显示,C. indicum(南京)(0.866)、C. rhombifolium(0.853)、C. indicum(天柱山)(0.847)、C. indicum(湖北)(0.844)、C. dichrum(0.841)和C. potentilloides(0.833),这形成了一个独立的支系,也展示了与栽培菊花更高的基因组相似性,比其他野生菊花物种更为接近(图3c和Supplementary Data 6)。值得注意的是,不同地理区域和染色体倍性水平的C. indicum存取被分为三个主要支系,并展示了比物种间观察到的还要多的遗传多样性(图3a),这在其他研究中也有发现。我们推测,菊花属内的种间杂交以及C. indicum栖息地多样性可能促成了这种遗传变异。

a 使用向日葵(H. annuus)作为外群的菊花属植物的最大似然系统发育树。分支上的数字表示支持值。菊花 cv. ‘钟山紫桂’和12种重新测序的二倍体或四倍体野生菊花物种的形态和细胞学特征在右侧展示。b UpSet图显示覆盖深度大于4×的100 kb非重叠滑动窗口的数量。c 12种野生菊花物种的相同得分(IS)值分布。更高的IS值表示与菊花的关系更为密切。每个箱形图的中心线是中位数,顶部和底部边缘对应于第一和第三四分位数,须表示从箱体边缘延伸出的1.5倍四分位范围。灰色点是异常值。每个物种的平均IS值以橙色点表示,并在箱形图顶部标记。数据使用单向ANOVA分析,随后进行双尾Tukey的诚实显著差异(HSD)多重比较测试(n = 70,524),不同的小写字母表示在P < 0.01的水平上统计显著差异。d 基于kmer的菊花 Smudgeplots分析。
为了进一步追踪渐渗事件,我们计算了菊花基因组上每个100 kb滑动窗口内的平均IS值。结果发现,栽培菊花中观察到大量小热点具有更高的IS值和少数大热点源自这些野生菊花物种,这表明广泛且多次的基因组渐渗可能有助于栽培菊花的形成以及菊花属物种复杂的网状进化历史。
C. nankingense长期以来被怀疑是栽培菊花的二倍体祖先。这项研究中组装的两个基因组使我们能够研究菊花三倍体化后的基因丢失和保留。C. nankingense中大约74.44%的所有基因与菊花中至少一个同源拷贝具有同源性,而55.10%的基因在菊花中保留了三个同源拷贝。然而,我们无法在菊花基因组中识别一个明显归属于C. nankingense的主导染色体组,与异源多倍体草莓和高粱的情况不同,这表明C. nankingense可能不是栽培菊花的直接祖先供体。这一发现得到了基于SNP的系统发育树(图3a)和IS分析(图3c)的支持。利用C. seticuspe基因组的组装,我们进行了类似的基因保留分析,发现C. seticuspe与菊花的关系不如C. nankingense密切。
栽培菊花是自体多倍体还是异源多倍体的起源仍不清楚。区分不同形式的多倍体并非总是直接的。通常,染色体行为、生育力、分离比率和形态学,结合遗传数据,是区分自体多倍体和异源多倍体的主要标准。以前的细胞学研究揭示,六倍体栽培菊花在其中二价体占主导地位的二倍体样的减数分裂中,但很少观察到单价体和多价体(主要是四价体)。然而,菊花的二态或多态遗传代表了一个连续体内的极端情况。我们在花粉母细胞(PMC)减数分裂期间进行的核型分析显示,18条染色体形成了9个二价体,而9条染色体保持为单价体(9II + 9I),而秋水仙碱诱导的双倍单倍体表现得像异源多倍体,几乎专门形成二价体(27 II)。基于唯一SNPs的每个同源组的系统发育分析显示,同一组内的三个同源染色体都彼此分离,除了Chr17和Chr18,这与菊花可能有异源多倍体起源的可能性一致。
尽管如此,我们发现与菊花基因组比较时,不同野生物种的覆盖均匀分布,这与已知的异源四倍体烟草和棉花的观察结果不同。这种不一致可能是因为菊花属内的物种关系密切;现存的二倍体或四倍体野生菊花物种可能也经历了广泛的网状进化
和高异质性;栽培菊花的假定原始祖先可能已经灭绝。为了验证这些结果不是由于选定的潜在祖先引起的,我们还使用了基于TEs分布的13-mer聚类方法,这不需要访问或甚至知道祖先谱系的活代表。尽管基于识别的4719个染色体特异性13-mer,每组三个同源染色体明显基于聚类,但我们发现所有同源染色体之间的13-mer计数除了Chr7-Chr8-Chr9外都存在明显差异,表明菊花可能不是严格的自体多倍体。此外,结果表明与自体多倍体紫花苜蓿相比,深度>120x的17-mer占据了更大的比例;菊花与C. nankingense之间的同源直系同源基因Ks分布观察到四个九个同源组中的四个存在显著差异;稍微不同的同源组内基因保留模式进一步支持了菊花可能不是严格的自体多倍体的可能性。有趣的是,Smudgeplot分析暗示菊花的基因组结构可能是“AAB”或“AB”。通过荧光原位杂交(FISH)分析比较三联体内的同源染色体也显示,两个同源染色体通常与第三个更为相似,但也呈现出差异,支持菊花中同源染色体的“AA’B”关系。
根据这些发现以及以前报告的证据,以及与其他典型的异源多倍体和自体多倍体的比较,我们得出结论,菊花 cv. ‘钟山紫桂’很可能是一个特定的片段异源多倍体。
菊花中同源基因表达偏倚
为了探索菊花的转录行为,我们比较了不同器官中注释基因的全基因组转录水平(Supplementary Note 6)。每个同源组在器官中的表达模式相似,除了位于第13号染色体中间的基因表达水平显著低于第14和第15号染色体上的基因(Supplementary Figs. 21 和 22)。我们关注了在三个同源染色体上的同源区块中显示1:1:1对应关系的11,438个基因,称为三联体。结合所有九个器官的数据进行的全局分析显示,21.65%的同源三联体表达没有统计学上的显著差异,被归类为平衡类别,从Chr1-Chr2-Chr3染色体组的19.20%到Chr22-Chr23-Chr24染色体组的23.50%(Supplementary Fig. 23);具有单一同源优势的同源三联体比例(41.73%)显著高于具有单一同源抑制的比例(36.61%)(Supplementary Fig. 24),其中Chr8优势占最大比例(24.7%)。GO分析指出,平衡三联体在九个同源组中丰富于不同的生物过程,这些过程与各种生命过程相关。特别是,‘蛋白稳定’、‘生殖结构发展’、‘多细胞生物发展的调节’、‘转移酶活性的调节’、‘叶发育’、‘射击系统发展’等,在两个同源组中被富集,而生长素运输(GO:0060918)出现在三个同源组中(Supplementary Data 9)。此外,我们发现同源基因方向和差异基因表达之间的相关性较弱(Supplementary Note 6, Supplementary Tables 15 和 16),这表明菊花中的同源基因表达偏倚可能不是由基因方向变化引起的。这些观察可能代表了菊花基因新功能化或亚功能化的第一步。
花朵进化和发展的遗传基础
花形在栽培菊花改良过程中经受了密集的选择,并继续是育种计划的主要目标。栽培菊花具有比其他菊科物种更多样化的头状花序形态,这主要由彩色和多样形态的辐射花瓣决定。根据花冠管合并的程度,花瓣可以被划分为扁平型、勺状型和管状辐射花(Supplementary Fig. 25c),但花瓣多样性的遗传基础尚不清楚。先前的研究表明,花器官的身份由MADS-box基因调控,作为众所周知的ABCE模型的一部分。在这里,我们在菊花基因组中鉴定了所有的MADS-box(Supplementary Fig. 26 和 Supplementary Data 10)和ABCE基因(Supplementary Fig. 25a)。结果显示,如其多倍体性质所预期的,菊花基因组拥有比其他菊科植物和拟南芥更多的MADS-box基因,特别是SEP和SVP类。同源基因对的同源性分析、Ks分析和重复分析揭示,最近的WGT-2以及较小规模的重复事件促进了MADS-box家族成员的扩张(Supplementary Fig. 6a, b),导致菊花基因组中每个ABCE模型基因至少有三个非常相似的副本。与未重复的同源基因相比,重复基因可以经历放松的功能约束,允许它们在基因序列和表达模式上发散,并进行亚功能化或新功能化。如Supplementary Fig. 25a所示,PISTILLATA(PI)同源基因确实显示出不同的表达模式。一个副本(evm.model.scaffold_4253.83)在营养器官中表达水平较高,而其他成员在生殖器官中高度表达,暗示它们的功能可能经历了亚功能化。相比之下,9种品种盘状小花中的六个AGAMOUS(AG)同源基因的表达水平显著高于辐射花中相应基因的表达水平,表明AG在决定花器官身份方面的功能保守。基因表达分析还表明,MADS-box B类基因PI和DEFICIENS(DEF)/APETALA3(AP3)可能在花瓣和雄蕊发育中起重要作用。
TCP家族CYCLOIDEA2(CYC2)类基因调节花的对称性、花序结构和生殖器官发育。先前的研究表明,CYC2类基因调节菊科物种如向日葵、千里光(Senecio vulgaris)和非洲菊的辐射和盘状小花的分化。我们在菊花中鉴定了25个CYC2样基因(Supplementary Fig. 25b),这比C. nankingense和向日葵分别发现的七个和八个成员要多得多。系统发育分析显示,菊花的CYC2a样基因经历了几次重复事件;一个亚类中的基因在生殖器官中高度表达,而另一个亚类中的基因在营养器官中高度表达,与其他CYC2类基因一致(Supplementary Fig. 25b)。先前的研究表明,CmCYC2c在C. lavandulifolium中的构成性表达导致辐射小花数量和辐射小花花瓣舌片长度的增加。然而,它们的表达与花瓣融合程度之间没有相关性,表明花瓣融合不仅由CYC2s控制,还由菊花中的其他基因控制。
为了进一步确定花瓣形状变异的分子遗传基础,我们在一个来自扁平型品种和管状型品种的F1群体中执行了全基因组混合分离分析测序(BSA-seq)方案(图4a, Supplementary Fig. 27 和 Supplementary Note 7)。使用ΔSNP-index和ED算法检测到与花瓣融合程度相关的72个一致的基因组区域(Supplementary Data 11)。特别是,作为CYC/DICHOTOMA下游、与DIVARICATA(DIV)对立控制花的对称性的MYB类基因RAD6(evm.model.scaffold_2712.53)被发现在qPT5-5(图4b)。该QTL在BSA-seq方法中一致检测到,位于第5号染色体的201.00-202.10 Mb区间内,对于SNPs的ΔSNP指数和ED峰值分别为0.91和1.65,对于InDels为0.76和1.15(Supplementary Fig. 28)。CmRAD6在扁平型品种中高度表达,并且是与花瓣融合程度负相关的turquoise模块中的一个中心基因(r = -0.92; P = 5.0E-04)(图4c, d 和 Supplementary Fig. 29)。此外,通过BSA-seq和加权相关网络分析(WGCNA)鉴定了几个花发育候选基因,如CYC2s、AGL6、MADS33(AGL12)、IBL1、PI(图4d 和 Supplementary Fig. 28)。还鉴定了几个生长素响应基因(Supplementary Data 12),表明生长素可能在花瓣发育中起关键作用,与我们之前的发现一致。这些结果提供了栽培菊花花瓣形状遗传结构的见解,而在此组装的参考基因组为加速未来QTL精细定位和育种计划提供了重要资源。
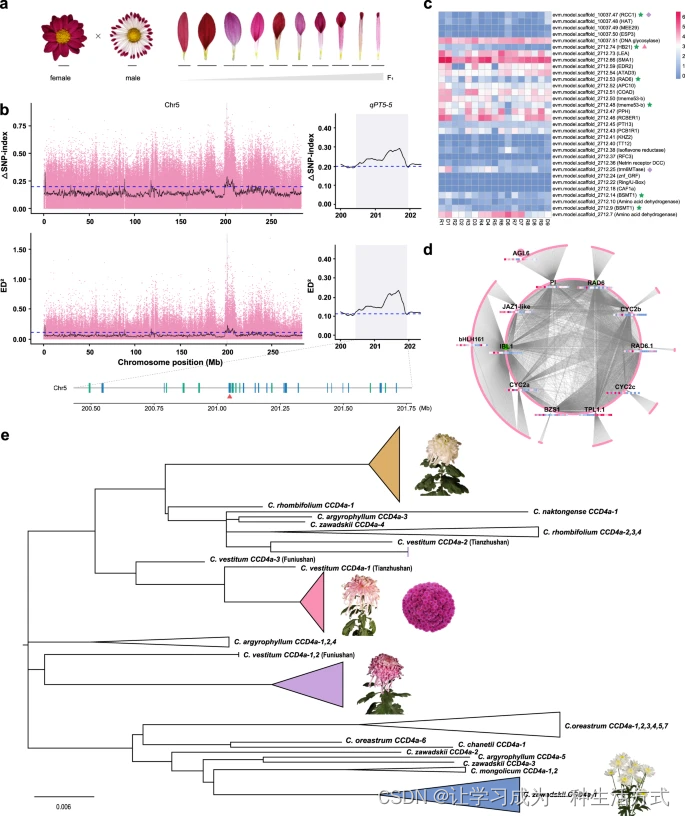
a 父母和后代之间花冠管融合程度的变异。标尺 = 1 cm。b 通过SNP-index(顶部)和欧几里得距离(底部)算法使用SNPs检测到第5号染色体上控制花瓣类型的QTLs。顶部1%的阈值由水平蓝色虚线指示。一个被指定为qPT5-5的显著峰,包含32个基因,在右侧部分放大并阴影显示。绿线和蓝线分别代表位于正义链和反义链上的基因。CmRAD6(evm.model.scaffold_2712.53)由红色三角形表示。c 三种扁平花瓣型栽培品种(R/D13)、三种管状花瓣型栽培品种(R/D46)和三种勺状花瓣型栽培品种(R/D7~9)的辐射(R)和盘状(D)小花中32个基因的表达。绿星、粉红三角形和紫色菱形分别代表在扁平与管状、扁平与勺状、管状与勺状组别中基因的差异表达。d 绿松石模块中基因的子网络。中心基因的热图与c面板相同。e 代表性非黄色野生和栽培菊花中CCD4a基因的系统发育树,展示了花色育种过程的潜力。橙色三角形合并了所有传统的日本菊花和一个传统的中国菊花栽培品种。粉红色三角形代表了传统的中国菊花和盆栽菊花的混合。紫色和蓝色三角形分别代表传统的中国菊花和切花菊花的组。完整树状图见Supplementary Fig. 31。
CmCCD4a的系统发育分析揭示了菊花花色育种的历史
栽培菊花的花色多样性主要是由于其彩色的辐射花瓣,这是这些植物的主要观赏性状之一。根据唐代(618-907)的文献,栽培菊花只有黄色的辐射花瓣,而宋代(960-1279)的文献记录了栽培菊花中各种颜色的辐射花瓣。一个重要的问题是,栽培菊花的花瓣颜色是如何从仅黄色变化为多种花色的。已经表明,类胡萝卜素裂解双加氧酶(CmCCD4a)调节菊花辐射花瓣中黄色素(类胡萝卜素)的降解(Supplementary Note 8 和 Supplementary Fig. 30c, e)。我们发现CCD4a在菊花基因组中缺失。此外,原始菊花品种‘钟山紫桂’具有紫色的辐射花瓣(Supplementary Fig. 1a)。然而,测序的单倍体材料显示黄色或略微橙色的辐射花瓣(图3a),并且通过人工全基因组加倍不能恢复花色(Supplementary Fig. 1b, c)。因此,我们假设野生菊花属物种的CCD4a基因通过人工杂交引入到栽培菊花中,但拷贝数较低,杂交后容易丢失,导致辐射花瓣恢复为黄色。
为了验证这一假设并确定CCD4a是如何被引入栽培菊花的,我们试图扩增40种栽培菊花品种和14种与菊花属相关的野生物种的CCD4a基因。结果显示,黄色或橙色辐射花瓣的栽培品种和野生亲缘中没有CCD4a基因。最终,我们从23种栽培菊花品种和9种菊花属的野生亲缘中克隆了78个CCD4a序列。系统发育分析显示,CCD4a通过多个独立的杂交事件被引入到不同的菊花栽培品种群中(图4e 和 Supplementary Fig. 31)。传统中国菊花和盆栽菊花的CCD4a基因可能来自C. vestitum,而切花菊花的CCD4a基因可能来自C. zawadskii(图4e)。这意味着,与传统菊花和盆栽菊花相比,切花菊花的育种过程涉及了不同的人工杂交事件。有趣的是,传统日本菊花的CCD4a聚成一个独立的支系(Supplementary Fig. 31),这表明菊花的花色性状可能源自日本的独立育种过程。总的来说,通过人工杂交多次引入CCD4a基因促进了原始菊花辐射花瓣中类胡萝卜素的降解,为现代栽培菊花的花色多样性做出了贡献。这一发现也支持了菊花的育种和传播历史。
讨论
由于高序列相似性和同源异源交换,多倍体基因组的组装和分析仍然是一个巨大的技术挑战。为了克服这些困难,我们选择了一个特定的单倍体栽培菊花作为测序材料,这大大降低了组装、注释和下游分析的复杂性。PacBio长读取测序允许解决和组装富含重复和高基因组相似性的区域。生成的菊花基因组具有1.87 Mb的contig N50大小、303.69 Mb的scaffold N50大小和27.99的LAI。它为进化和比较基因组学研究提供了宝贵的资源。我们的数据表明,先前报道的C. nankingense的最近WGD事件,估计约在6百万年前,更可能是一个WGT事件(在此称为WGT-2),并且由所有菊花属物种共享(图2b和Supplementary Fig. 6)。此外,还揭示了栽培菊花中一个非常近期的独立三倍体化事件(约3百万年前)(图2b)。因此,菊花在WGT-γ之后总共经历了至少三轮WGT,加上最近的LTR爆发(<0.5百万年前;图2d),导致了庞大、复杂的基因组结构。
多倍体长期被认为是植物进化和物种形成的主要力量。迄今为止,已经测序了七种六倍体植物的基因组,所有这些基因组都被认为是异源多倍体的起源。先前的研究揭示,盆栽菊花在遗传上与野生菊花祖先物种的关系比切花菊花或传统菊花更为密切。因此,‘钟山紫桂’基因组揭示了栽培菊花的起源。基于我们当前的发现以及先前的报告,我们推断,多个已识别的渐渗、杂交和多倍体化事件导致了菊花多倍体复合体的进化网状,并且先前认为是栽培菊花的祖先的C. nankingense、C. lavandulifolium和C. seticuspe,可能不是直接的祖先供体。系统发育和IS分析(图3)证明了在巫山(重庆,中国)狭窄分布的C. rhombifolium与栽培菊花关系更为密切。还有待进一步探索的问题包括C. rhombifolium在栽培菊花形成中的可能涉及,以及杂交混合物何时以及如何扩大其范围。我们在这里提供的多线证据结合以前的研究,暗示菊花 cv. ‘钟山紫桂’很可能是一个特定的片段异源多倍体(按照Stebbins等人的定义)并揭示了其正在进行的发散进化。不幸的是,我们未能将27个拟染色体分配给亚基因组,我们关于菊花“AA’B”基因组构成的猜想仍需进一步研究。有趣的是,我们发现只有约22%的菊花同源基因显示平衡表达,这远低于异源多倍体小麦(72.5%)和芸苔属植物Brassica juncea(83.8%)的百分比。这可能由栽培菊花的快速进化和其特定的无性繁殖解释。
栽培菊花比其他菊花属物种具有更大的形态多样性,特别是在花形和颜色方面。在这项研究中,我们推断回溯至6百万年前的时间段是菊花种化和栽培菊花快速进化的重要时期。最近的WGT-2和三倍体化事件可能导致了花形态多样性的扩张。基于参考基因组,我们能够初步揭示栽培菊花遗传多样性的变异底层(Supplementary Data 13)并识别了几个涉及花瓣发育的候选基因,这将允许进一步的精细定位和功能验证。此外,我们对CCD4a的系统发育研究提供了每种栽培类型甚至不同栽培品种独立育种历史的视角。此外,我们重建了菊花花色化的花青素和黄酮醇生物合成途径(Supplementary Note 8 和 Supplementary Fig. 30d)。
总之,我们在这里提供的基因组资源可以帮助挖掘控制菊花重要性状的中心基因,并有助于研究菊花的进化。此外,参考基因组将促进分子标记辅助育种和基因组编辑在栽培菊花中的应用。菊花的异源多倍体基因组可能有助于指导其他具有高异质性和不确定起源的大型复杂多倍体基因组的组装。
方法
植物材料和基因组测序
我们对之前表征的盆栽和地栽型菊花 cv.‘钟山紫桂’(2n = 6x = 54)的一株单倍体(n = 3x = 27)进行了整个基因组的测序,该株单倍体是通过非受精卵母细胞离体培养获得的(Supplementary Fig. 1)。从幼嫩叶片组织中提取基因组DNA使用DNAsecure Plant Kit(TIANGEN,中国北京)进行Illumina短读测序、PacBio长读测序、10X Genomics和Hi-C文库构建和测序,详见Supplementary Note 1。为了支持基因组注释,从各种器官中提取总RNA使用RNA提取试剂盒(华粤洋,中国北京)进行测序(RNAseq)。本研究使用的所有植物材料均来自菊花种质资源保藏中心(中国江苏南京,南京农业大学)。
染色体组装和验证
利用PacBio SMRT测序仪获得的长读序列进行了长读组装,采用FALCON75进行了全新组装,参数设置如下:length_cutoff_pr = 11,000,overlap_filtering_setting = --max_diff 500 --max_cov 500。根据PacBio长读序列,使用Quiver76对初始组装进行了修正。然后,使用默认设置的BWA-MEM77将10X Genomics读序列与组装进行了比对。使用条形码测序读数进行了FragScaff78进行脚手架构建。利用Illumina序列进行了错误校正,使用Pilon79进行了基于Illumina序列的校正。随后,使用BWA-MEM4将Hi-C测序读数与组装的脚手架比对,并使用ALLHiC80对这些脚手架进行了聚类和重排序。利用Juicebox81对脚手架进行了微调,从脚手架中移除了不一致的contigs,经过仔细的手动检查后生成了最终的染色体组装。将前10×最长的PacBio读序列使用minimap2(参数:“-k 15 -w 10 -I 9 G”)进行了映射,以计算其连续性,方法是计算唯一映射(≥80%的对齐长度)比例。此外,我们还根据Supplementary Note 2的描述,为二倍体C. nankingense进行了染色体尺度的基因组组装。使用MCScan(Python-jcvi)对二倍体C. nankingense和菊花的基因组进行了同源性分析。
使用Embryophyta odb10数据库的CEGMA (http://korflab.ucdavis.edu/dataseda/cegma/)82、BUSCO (http://busco.ezlab.org/)83以及LTR Assembly Index (LAI)84来评估基因组组装的完整性和连续性。利用BLAT (Blat Spec and User's Guide)将ESTs映射到组装上,评估基因完整性。
基因组注释
我们采用了蛋白质同源、全新和转录组数据相结合的预测方法来注释蛋白质编码基因。使用TBLASTN25将六种物种(拟南芥、胡萝卜、向日葵、莴苣、番茄和马铃薯)的蛋白序列与菊花基因组进行比对,E值设定为1e-5。使用GeneWise84预测每个BLAST命中区域的准确基因结构。对于基于转录组的预测,使用TopHat(--splice-mismatches 2 --max-intron-length 500000 --min-intron-length 50)和Cufflinks85(--max-intron-length 500000 --min-intron-length 50 --max-mle-iterations 5000)将三种组织的RNA-seq数据映射到菊花基因组上。此外,使用Trinity86组装RNA-seq数据(--min_glue 2 --min_kmer_cov 2),然后使用PASA软件87改进基因结构。生成的基因集被标记为PASA-T-set,并用于训练全新基因预测程序。使用五个全新基因预测程序,即Augustus(版本2.5.5)88、GENSCAN(版本1.0)89、GlimmerHMM(版本3.0.1)90、Geneid91和SNAP92,预测了在重复遮蔽的基因组中的编码区域。最后,使用EVidenceModeller (EVM)93将所有预测组合起来生成非冗余的基因集。
蛋白质编码基因的功能注释通过BLASTp搜索(E值 ≤ 1e-5)94针对SwissProt (UniProtKB/Swiss-Prot)、NR (ftp://ftp.ncbi.nih.gov/blast/db/)、InterPro (InterPro, V32.0)、Pfam (Pfam is now hosted by InterPro, V27.0)和KEGG数据库 (KEGG Database)进行。
通过多种数据库和软件包,包括RepeatModeler95、LTR_FINDER96、RepeatMasker和RepeatProteinMask97的全新和同源性方法,识别了菊花基因组中的转座元件(TEs),详见Supplementary Note 2。LTR反转录转座子使用一个管道进行注释,该管道使用LTRharvest98、LTR_FINDER96和LTRdigest99。我们提取了每个LTR的长末端重复序列,使用MUSCLE100对其进行了比对,然后用Kimura二参数方法计算LTR之间的距离(K)。LTR插入时间估计使用下式进行:T=K/(2×r),其中r是Asteraceae家族的一般替代率,为1.3 × 10−8每个位点每年。额外细节见Supplementary Note 3。
基因家族鉴定
使用拟南芥中ABCE花发育基因的蛋白序列和菊花中报道的CYC2基因作为查询,在菊花参考基因组中使用BLASTp进行识别,设置阈值为E值 ≤ 1e-5。选择至少覆盖种子蛋白序列50%以上且蛋白序列相似性 >50%的对齐结果作为同源物。然后,使用PFAM(Pfam is now hosted by InterPro)预测这些同源物的结构域。只考虑具有相同蛋白质结构域的基因为同源物。提供了在包括菊花、A. annua、C. cardunculus、C. nankingense、C. seticuspe、L. sativa、H. annuus和A. thaliana在内的八种植物物种中鉴定TPS和MADS-box家族成员的结果,详见Supplementary Note 3。
栽培菊花的起源
对于全基因组重测序,选择了12个中国野生菊花种质,代表了所有现存的潜在的二倍体和四倍体栽培菊花的祖先。根据经过过滤的SNP,通过构建最大似然树和计算相同得分(IS)值来评估系统发育关系。使用染色体级别的C. nankingense和C. seticuspe基因组作为参考进行了基因保留分析。为进一步澄清菊花的基因组结构,采用了两种基于kmer的方法,并检查了测序的单倍体和倍性植物的减数分裂行为。进行了FISH分析以评估同源染色体之间的序列差异。所有细节见Supplementary Note 5。
挖掘花形状的数量性状位点和候选基因
基于参考基因组,我们能够通过整合BSA-seq和WGCNA来识别数量性状位点和候选基因,详见Supplementary Note 7。简而言之,对于BSA-seq分析,在‘红小’和‘Q5-12’的F1群体中构建了两个极端DNA池,即扁平体积(BF)和管状体积(BT),每个池中混合了20个个体的DNA,详见Supplementary Fig. 25。对于四个文库,包括两个父本和两个子代池,使用DNBseq-T7平台测序以获得平均覆盖率为30×的150 bp PE reads。使用GATK软件进行了SNP/InDel变异调用。使用了两种关联分析方法,SNP-index和欧氏距离(ED)。绝对ΔSNP指数和ED值的前1%被认为与花冠管合并度(CTMD)强相关。
对于WGCNA,在早期开花阶段分别对九个代表性品种的花瓣和盘瓣进行了解剖,提取RNA进行转录组测序。使用DESeq进行了差异基因表达分析。在过滤了样本中低丰度(FPKM ≤ 1)的基因后,使用74,074个花瓣特异基因进行了WGCNA软件包的共表达网络分析,使用最小模块大小为50个基因和合并阈值为0.25的动态树切割算法。将CTMD值用作表型数据来识别与花瓣类型相关的模块。共表达网络在Cytoscape软件中可视化。
CCD4a基因的系统发育分析
为了获得菊花属物种的CCD4a核苷酸序列,设计了基于CCD4a开放阅读框(ORF)序列的引物对CCD4a-ORF-F(5'-ATGGGCTCTTTTCCCACATCT-3')/R(5'-ATAATCAAAGCGTTGTTAGGTCATT-3')。PCR混合物包括17μL ddH2O,25μL 2× Phanta Max缓冲液,1μL dNTP混合物,2μL每个CCD4a-ORF-F/R(Sangon Biotech,中国上海),1μL Phanta Max高保真度DNA聚合酶(Vazyme Biotech,中国南京),和2μL DNA。PCR程序包括95°C 3分钟,然后进行35个循环:95°C 15秒,55°C 15秒,72°C 120秒,最后延伸72°C 5分钟。将PCR产物插入pEASY-Blunt Simple Cloning载体(TransGen Biotech,中国北京),然后转化为DH5α进行测序。使用MUSCLE软件进行多序列比对,使用MEGA X构建ML方法111的系统发育树。内部支持值是使用1000次Bootstrap重复估计的。