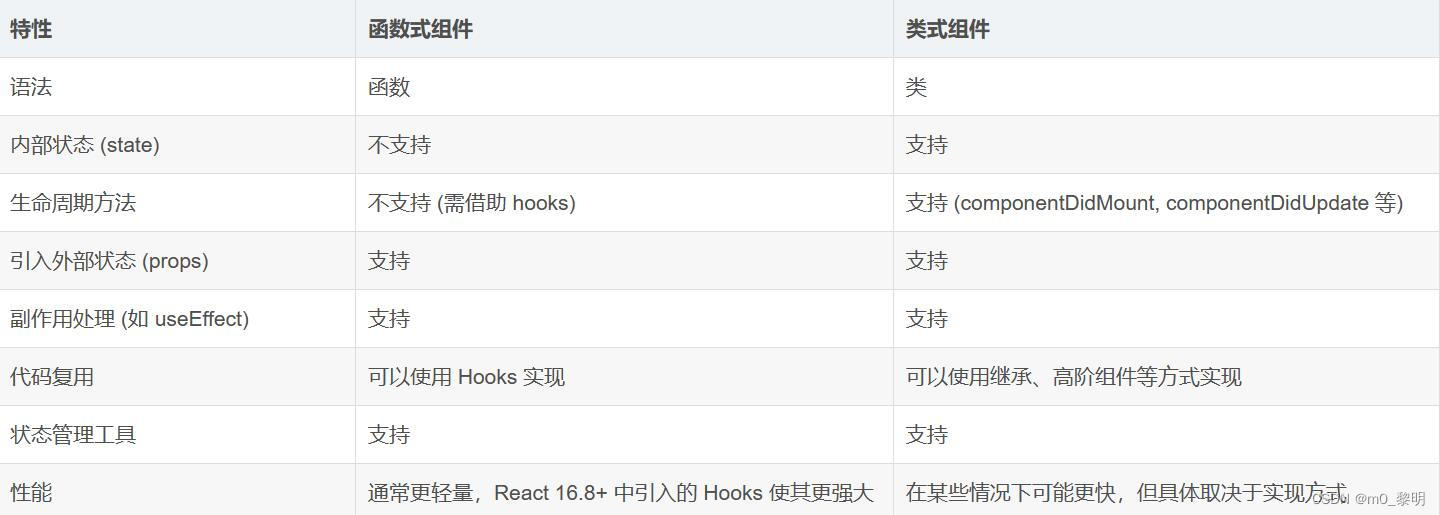
这里我们分享一个热图函数,是一个可视化的函数,您只需要提供作图的matrix数据即可。最近对于图形注释使用点比较上头,所以我们这个函数的列注释使用设置的·是点的注释形状。我们的函数解决了以下几个问题:
-
数据的自动排序和列的自定义排列,数据会按照你需要的列顺序自动从大到小排列,让热图可视化更加美观。
-
热图的列自动注释,我们采用了点的注释。
-
legend的修改,包括刻度等修饰。
-
行的注释,行的注释需要自行 调整,函数设置了可选择选项。
-
行的标签,在函数中我们设置了如果矩阵nrow行数大于100自动不显示行名,应为那样都挤在一起也没啥用。后续可自 行挑选需要的行进行展示。
-
需要注意一下,函数有一个参数data_scale,假设你的数据不需要scale标准化,那么参数选择T,作图使用你的原始数 据。如果需要scale标准化,那么选择F,会对你的数据进行标准化处理。
看一下函数参数:(未标注参数与Complexheatmap::Heatmap一致)

参考链接:
marker基因注释热图可视化函数(视频教程-通用函数)_哔哩哔哩_bilibili
接下来我们看看函数具体的使用,首先我们用一个ATAC TF分析的数据,这个矩阵是已经导出的,行是celltype,列是TF。其 实就是我们平时做热图的时候一样的数据。Order_col就是设置列的顺序,我这里直接演示就使用了数据的列名排列,如果 需要自己设置,这里传入一个向量,设置顺序即可。颜色也是可以设置的。
setwd("D:/KS项目/公众号文章/scATAC-scRNA marker基因注释热图")
#=================================================================================
#---------------------------------------------------------------------------------
TF <- c("JUND (264)","FOSB (190)","JDP2 (178)","TAL1 (42)","TAL2 (42)","TCF21 (21)","TCF23 (21)","BCL11A (799)","BCL11B (799)","ELF2 (364)","NFIX (89)","GBX2 (87)","SOX9 (145)","SOX4 (95)","SOX13 (92)","SOX12 (91)","NHLH2 (149)","TCF12 (159)","ZBTB7A (98)")plot_atac <- read.csv('plot_atac.csv', header = T, row.names = 1)pdf('heatmap1.pdf', width=4, height=6)
heata = ks_Heatmap(mat = plot_atac,data_scale = T,Order_col = colnames(plot_atac),legendTitle = "Norm.Enrichment -log10(P-adj)[0-Max]",point_size = 5,heat_col = c("#E6E7E8","#3A97FF","#8816A7","black"),TitlePosition = "leftcenter-rot",legend_at = c(0,50,100),legend_Lab = c(0,50,100),column_names_gp = gpar(fontsize = 6),row_names_gp = gpar(fontsize = 6),customRowLabel=TF,colanno_color = c("#7DD06F","#844081","#688EC1","#C17E73","#484125","#6CD3A7","#597873",'#3361A5'),rowAnn = T,rowAnno_num = c(16,3,1,4,12,9,8,11))draw(heata, merge_legends = TRUE,heatmap_legend_side = "right")
dev.off()

接下来我们用单细胞转录组的数据进行演示:我们这里的演示选择的是每个celltype的top10marker基因。首先计算 marker基因,并计算每个celltype的平均值。获得作图数据。
library(Seurat)
library(dplyr)
#单细胞数据的演示\先找marker基因
DefaultAssay(sce) <- "RNA"
Idents(sce) <- "celltype"
all.markers <- FindAllMarkers(sce, only.pos = TRUE, min.pct = 0.5, logfc.threshold = 0.5)
top10 <- all.markers %>% group_by(cluster) %>% top_n(n=10, wt=avg_log2FC)
table(top10$cluster)
# SMC LY UEC SF CEC EC MAC
# 10 10 10 10 10 10 10
#计算平均表达量
gene_cell_exp <- AverageExpression(sce,features = top10$gene,group.by = 'celltype',slot = 'data')
gene_cell_exp <- as.data.frame(gene_cell_exp$RNA)
作图:
genes = c("ACTA2","ADIRF","MYL9","TPM2","CCL4","CCL5","NKG7","CD96","RHEX","NPAS3","SFRP4","COL3A1","COL1A1","IGF1","RORB","CAPS","CFAP299","CFAP47","RSPH1","DNAH11","PTPRB","INSR","ENPP2","IL1B","HLA-DQA1","HLA-DRA","HLA-DPA1","HLA-DRB1","HLA-DPB1","CXCL8")#plot
pdf('heatmap2.pdf', width=4, height=6)
heata = ks_Heatmap(mat = gene_cell_exp,data_scale = F,Order_col = colnames(gene_cell_exp),legendTitle = "Expression",point_size = 5,heat_col = c('#e0f3db','#a8ddb5','#4eb3d3','#0868ac','#084081'),TitlePosition = "leftcenter-rot",legend_at = c(-1,0,1,2,3),legend_Lab = c(-1,0,1,2,3),column_names_gp = gpar(fontsize = 6),row_names_gp = gpar(fontsize = 6),customRowLabel=genes,colanno_color = c("#7DD06F","#844081","#688EC1","#C17E73","#484125","#6CD3A7","#597873"),rowAnn = T,rowAnno_num = c(10,10,10,10,10,10,10))draw(heata, merge_legends = TRUE,heatmap_legend_side = "right")
dev.off()

这里我们可能会发现一个问题,明明是每个celltype10个gene,为什么行注释好像不是很对,这是应为有些基因不仅在一 中celltype中高表达,而数据是按照表达从高到低排序的,所以才会出现这个问题,可以自行调整注释的数目,或者不采用注释等。一个函数不可能完成所有的事情,但是我们提供了框架和思路,可自行修改拓展。觉得分享有用的点个赞,分享一下再走呗!




![【QT5-程序控制电源-[GPIB-USB-HS]-SCPI协议-上位机-基础样例【2】】](https://img-blog.csdnimg.cn/bddbb4017bb14b49b5ed2915f3e8cf7c.png)