Title:PeSTo: parameter-free geometric deep learning for accurate prediction of protein binding interfaces
期刊:nature communication
分区:一区
影响因子:16.6
webserver:t Pesto
Github:GitHub - LBM-EPFL/PeSTo
摘要
蛋白质是生命的重要分子组成部分,由于其特定的分子相互作用,负责大多数生物功能。然而,预测它们的绑定接口仍然是一个挑战。在这项研究中,我们提出了一个几何变换器,它直接作用于仅标有元素名称的原子坐标。由此产生的模型——蛋白质结构转换器,PeSTo——在预测蛋白质-蛋白质界面方面超越了当前的技术水平,并且还可以高度自信地预测和区分涉及核酸、脂质、离子和小分子的界面。它的低计算成本使得能够处理大量的结构数据,例如分子动力学系综,允许发现在静态实验解决的结构中保持不明显的界面。此外,由从头结构预测提供的不断增长的折叠体可以很容易地被分析,为揭示未探索的生物学提供了新的机会。
方法和数据集
数据集
数据集由来自蛋白质数据库的所有生物组件组成。使用簇之间最大30%的序列同一性来聚集亚基。亚基簇被分成大约70%的训练集(376216条链),15%的验证集(101700条链),和15%的测试集(97424条链)。我们通过评估验证集上的模型来选择最佳超参数。测试集由包含来自MaSIF-site基准数据集的53个亚单位或来自蛋白质-蛋白质对接基准5.038 (PPDB5)数据集的230个结构中的任何一个的簇组成。此外,我们提取了在ScanNet15的基准数据集和PeSTo的测试数据集中常见的子集417结构。除非特别说明,所有被选择用来评估模型预测质量的例子都属于测试集。
特征和标签
我们确定了PDB上30种最常见的原子元素。元素被用作onehot编码。输入矢量要素最初设置为零。距离矩阵和归一化位移向量矩阵被用作几何特征。氨基酸、核酸、离子、配体和脂质分别选自20、8、16、31和4种最常见的分子。用于帮助解决结构的非天然分子被忽略。界面被定义为5英寸以内的残基-残基接触。所有蛋白质-蛋白质界面以及蛋白质-核酸、蛋白质-离子、蛋白质-配体和蛋白质-脂质界面都被识别。每个子单元的接口细节作为交互类型矩阵(79×79)存储在数据集中。这使得能够在训练会话开始时选择特定接口作为标签,而不必重建整个数据集。界面靶标可以从79个可用分子的子集的任意组合中选择。
方法
蛋白质结构的Transformer架构
输入特征被嵌入到一个输入状态大小为S = 32的三层神经网络中,其中隐层大小为32。每个几何变换器由3层的5个神经网络组成,以执行补充算法1中描述的多头自关注(S = 32,Nkey = 3,Nhead = 2)。对于原子数量小于设置的最近邻(nn)数量的结构,额外的不存在的相互作用被发送到具有设
置为零的标量和矢量状态的汇聚节点。连续应用4组8个几何变换器,每组的最近邻数量递增(nn = 8、16、32和64)。如补充算法2中所述,通过在形成每个残基(S = 32,Nhead = 4)的原子上使用局部多头掩码,几何残基汇集模块将结构的原子级编码聚集成残基级描述。最后一个模块是一个多层感知器,具有3层S = 32的隐藏大小,解码所有残差的状态并计算预测,返回从0到1的置信度得分。
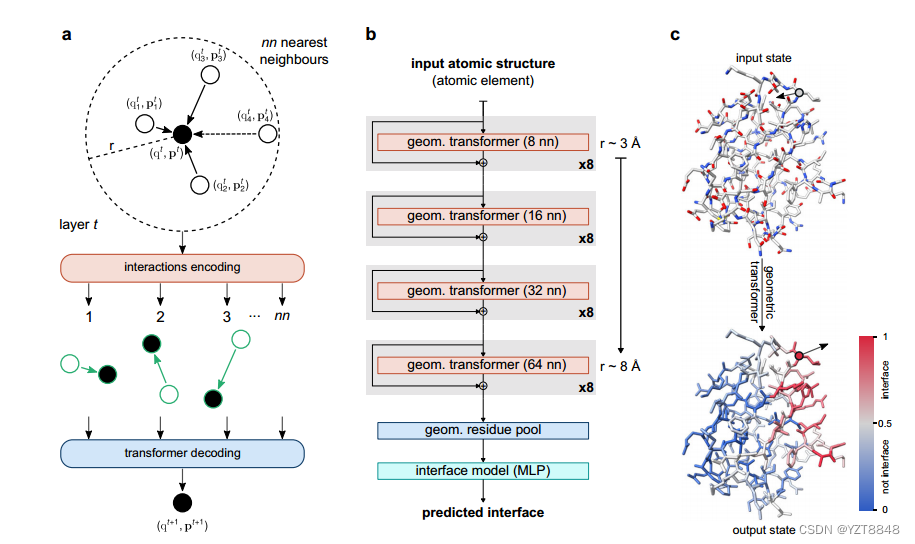
Transformer的蛋白质结构(PesTo)
许多成功的方法结合了transformers和几何深度学习,将结构表示为图形或点云,并整合了神经网络的不变性或等方差要求。主要突破来自蛋白质折叠领域,其中AlphaFold将注意力整合到Evoformer模块和结构模块中,RoseTTAFold20模型的第三个轨道使用阿瑟(3)转换器在折叠过程中细化原子坐标。此外,递归几何网络 (RGN2)利用Frenet-Serret公式来表示蛋白质的主链,几何矢量感知器 (GVP)使用线性运算来构成带有gating的矢量特征。已经开发了多种其他基于机器学习的蛋白质-蛋白质相互作用位点预测方法。
我们在这里介绍PeSTo,一个无参数的几何变换器,直接作用于蛋白质结构的原子。如图1所示以及在方法中详细描述的,该结构被表示为以原子位置为中心的点云,并且其几何形状通过保证平移不变性的成对距离和相对位移向量来描述。仅使用它们的元素名称和坐标来描述原子,而没有任何明确的数值参数化,例如质量、半径、电荷或疏水性。每个原子都与一个标量态(q)和一个矢量态(p)相关联,对结构的性质进行编码。如图1a所示,我们定义了作用于该点云的几何变换操作,以使用其局部邻域中的状态和几何来更新这些状态。所有最近邻(nn)的原子之间的相互作用使用几何形状(即,距离和位移向量)和所涉及的原子对的状态来编码。多头注意力层最终解码并调节信息的传播(补充算法1)
几何变换操作是平移不变的、旋转等变的,并且独立于原子的顺序和相互作用的顺序。为了保持向量状态的旋转等方差(参见补充方法),转换器注意力线性地组合来自局部几何形状和局部状态向量的缩放向量,以基于局部上下文动态地传播向量状态信息。注意操作允许动态数量的最近邻居(nn)。然而,在实践中,对于固定数量的最近邻,该操作的计算效率要高得多。与对图像应用卷积运算的方式相同,链接几何变换可以在比单个运算的局部上下文更远的范围内传播信息。因此,主要架构基于自下而上的方法,从8个最近邻居(≈3.4半径)的小环境开始,直到与64个最近邻居(≈8.2半径,图1b)的远程交互。
上下文的大小逐渐增加,允许模型逐渐包括更多的信息,同时保持深度模型的计算要求和存储器更便宜。几何变换器之间的剩余连接能够训练更深的神经网络结构。两个额外的模块在残基水平上聚集基于原子的几何描述,而不依赖于残基内的原子数(即几何残基汇集,补充算法2),并预测每个氨基酸是否在相互作用的界面上(图1c)。
与之前使用球谐函数对几何背景进行编码的SE(3)transformer31等方法相比,我们的方法仅使用向量,通过transformer attention调制其信息。与等变卷积相比,我们的方法基于具有几何图形的图,并且使用变换器来执行消息传递。
训练
该模型被训练来预测蛋白质与蛋白质、核酸、配体、离子或脂质的界面。最佳神经网络架构在单个NVIDIA V100 (32 GB) GPU上训练了8天。具有最多8192个原子(≈100 kDa)且不含氢的亚基用于限制训练期间的记忆需求。少于48个氨基酸的亚基在训练中被忽略。我们只在PDB数据库提供的第一个生物装配上进行训练。过滤后的有效广义蛋白质界面数据集由113805个用于训练的亚基和29786个用于测试的亚基组成。
结果
Protein–protein interface prediction
我们使用来自PDB的300,000多条蛋白质链训练了一个PeSTo模型(参见“方法”),以预测蛋白质-蛋白质界面中涉及哪些残基,其输出值范围从0到1(图2a)。零表示预测残基不会参与相互作用,而值1预测残基会在界面上。实际上,预测的实际值反映了残差水平上的预测的置信度,使得远离0.5的值意味着更高的置信度,参见补充图1。
我们首先评估了PeSTo的性能,并与解决类似任务的最新方法(即ScanNet15)进行了比较。我们使用了两种方法共有的417个结构的基准数据集(参见“方法”)。在该基准上,PeSTo优于没有多重序列比对(MSA)的ScanNet,具有0.93对0.87的中值接收操作特征(ROC)曲线下面积(AUC)(图2b和补充表1用于对不同数据集和指标的扩展比较,包括precisionrecall AUC和Matthews相关系数)。此外,我们对这两种方法的速度进行了定量比较(补充图2),发现PeSTo的平均运行时间(5.3±2.8秒)和不含MSA的ScanNet的平均运行时间(9.1±1.8秒)在CPU上不相上下。然而,具有MSA(160±83秒)的ScanNet的运行时间比PeSTo慢两个数量级,根据所有指标(补充表1),相对于PeSTo没有提供实质性的改进。
我们在用于基准测试MaSIF-site7,36(目前可用的最佳算法之一)的同一数据集上进一步比较了PeSTo,我们以30%的序列同一性将其从我们的训练集中排除。PeSTo达到0.92的曲线下中值接收操作特征(ROC)面积(AUC ),而MaSIF-site为0.8,接着是SPPIDER35和PSIVER37(图2b)。对于53个结构中的38个,PeSTo预测的界面具有比所有其他方法更高的ROC AUC。
最后,我们将PeSTo预测的蛋白质-蛋白质界面与alpha fold-多聚体预测的进行了比较。我们从PeSTo和AlphaFold(见“方法”)的验证集中的结构中选择了23个二聚体(即46个界面)。我们观察到PeSTo的表现几乎与α折叠多聚体一样好(见补充表2 ),而没有计算任何多序列比对的额外成本。因此,这些结果显示了我们的方法如何能够以与α折叠多聚体相当的准确度用于快速筛选潜在的界面。
为了进一步展示现实世界应用中的预测质量,我们测试了蛋白质-蛋白质对接基准5.038 (PPDB5)数据集的蛋白质的未结合构象。图2a中的实施例显示PeSTo从其未结合的构象(离结合状态0.93 RMSD)恢复了链菌素B与卵类粘蛋白的相互作用界面,ROC AUC为0.96。总的来说,在由各种不同难度的靶组成的整个PPDB5数据集上,对于蛋白质-蛋白质对接的一般任务,PeSTo对未结合结构的预测达到了0.78的ROC AUC中值,对各个结合状态的预测达到了0.85。
重要的是,运行该模型所需的时间很短(例如,对于100 kDa的蛋白质,在单个NVIDIA V100 GPU上从PDB加载到预测需要300 ms,补充图3),这使我们能够有效地评估从分子动力学(MD)模拟中提取的大型结构集合的快照。我们应用PeSTo对构象进行蛋白质-蛋白质界面预测,这些构象是通过对从PPDB5中提取的20个选择的二元复合物的实验性衍生的未结合和结合亚基进行1 s长原子MD模拟而采样的(图2c)。结合的和未结合的结构以及MD取样的构象分别达到0.85、0.82和0.79的ROC AUC中值(参见补充表3的其他指标)。
我们观察到该模型在实验解决的结合和非结合构象上表现几乎一样好。尽管总体上ROC AUC随着结合结构的RMSD增加而降低(补充图4),但对于大多数结构和MD取样构象,我们的方法仍然能够恢复ROC AUC高于80%的界面。
在某些情况下,用PeSTo处理未结合蛋白质的MD轨迹比在起始静态结构上运行PeSTo时更好地识别某些界面,这表明我们的方法在现实生活情况中的有效实际应用(图2d)。努力为PeSTo的日常应用提供一个协议,我们认为用户可能会寻找一些高等级的残基预测来表征结合界面。因此,我们将“回收率”定义为预测10%高级残基的能力,在我们的多维数据集的情况下,这相当于3 2个残基。如果所有这些残基预测正确,我们认为界面完全恢复。在由40个组成亚基和相关界面组成的20个复合物中,当直接应用于未结合亚基的实验结构时,该模型对16个界面具有完美的恢复率。在剩余的24个案例中,我们表明,使用MD更广泛地对蛋白质构象景观进行采样并聚类以进一步分组预测的界面,完全恢复另外16个亚单位(80%)的结合界面是可能的。
例如,PeSTo预测实验解决的未结合的猪胰腺弹性蛋白酶(PDB ID 9EST)的结构没有界面(图2e)。与弹力素结合的复合物(PDB ID 1FLE)相比,未结合的实验构象的主链RMSD为1.2。
然而,从单独的未结合的猪胰腺弹性蛋白酶开始的MD模拟显示构象转换导致与弹性蛋白的相互作用界面的恢复,具有0.92的簇中心ROC AUC和预测结合界面的完美恢复率(即,在这种情况下3个残基)。检查MD模拟揭示了弹性蛋白酶中的环的运动是允许弹性蛋白进入口袋并容纳分子间β-折叠所必需的,分子间β-折叠使复合物稳定,如实验所解决的。
通用蛋白质绑定界面预测
根据蛋白质-蛋白质界面预测的结果,我们扩展了该模型以发现和识别更多类型的界面,产生了一个广义的PeSTo模型,该模型预测蛋白质与其他蛋白质以及核酸、离子、配体和脂质的相互作用界面。我们训练了一个具有PDB结构的广义PeSTo模型,该模型具有所有类型的预期相互作用,如方法中所述。蛋白质-核酸界面的界面预测几乎与蛋白质-蛋白质界面一样好,测试组的ROC AUC达到0.89(图3a)。通用模型还可以检测离子、配体和脂质界面,ROC AUCs在每个测试集上分别为0.87、0.86和0.77(其他指标见补充表4)。该模型确实经历了离子和配体之间的一些混淆,如混淆矩阵所揭示的(补充图5)。蛋白质-脂质预测的较差性能取决于目前在PDB可获得的非常有限的蛋白质-脂质复合物数量(仅占我们汇编的可利用数据的0.7%)。我们注意到,在相同的数据集上重新训练模型,但是在训练、验证和测试集之间具有最大5%的序列同一性,而不是30%,导致在所有界面预测类型上平均在1% ROC AUC内的等效性能,证实了PeSTo在同源性降低上的稳定性。
接下来,我们举例说明了广义PeSTo模型,展示了测试集中的五个例子,这些例子证明了它在各种界面之间进行辨别的能力,即使当它们在PDB中重叠或代表性不足时。第一个例子(图3b)对应于大肠菌素E7核酸内切酶结构域,其通过包含锌离子的界面结合DNA(PDB ID 1 ZnS)。通过广义PeSTo运行脱辅基蛋白返回两个界面的正确预测,甚至在重叠部分。第二种情况(图3c)对应于由RUNX1形成的复合物,其一端结合有dsDNA,另一端结合有蛋白质CBFβ(PDB ID 1H9D)。
通过一般化模型运行分离的RUNX1通过DNA和蛋白质通道返回清晰、准确的界面。在第三个实施例中(图3d ),我们用结合RNA的抗体(PDB ID 6U8K)的结构来挑战一般化的模型,这与大多数可获得的结合其它蛋白靶标的抗体相反。广义模型正确地预测了蛋白质没有界面,而RNA有正确的界面。
尽管在与脂质的界面上,广义PeSTo表现不太好,但在实践中,我们观察到该模型能够准确地检测可溶性蛋白质的脂质结合袋(由图3e中的类固醇生成因子举例说明)甚至跨膜蛋白的膜结合区(补充图6)。
尽管没有经过专门的训练,但在这两种情况下,PeSTo都能够检测到具有更强得分的脂质的特定口袋。我们注意到,许多蛋白质与脂质的界面在PDB结构中仅部分明显(例如,单个脂质结合到膜扫描区域),导致低训练数据质量,从而导致ROC AUC的人为下降。
有趣的是,我们还发现PeSTo将其预测能力扩展到其自身的训练之上,例如DNA结合细菌整合宿主因子(mIHF)的情况,其DNA结合形式的X射线结构是可用的(图3f)。这种结构在生物装配中呈现一个DNA结合界面39,该界面包括在训练集中,但是溶液状态NMR滴定显示了更广泛的相互作用表面,主要分布在弯曲DNA所需的两个表面区域,如AFM40所示。PeSTo对这种蛋白质的预测超越了它的训练,指出了两个与溶液中核磁共振数据非常匹配的表面补丁。
人类蛋白质组结合界面的高通量预测
我们试图探索整个人类蛋白质组,并分析我们下文所称的界面组,即所有能够结合其他蛋白质、核酸、脂质、配体和离子的潜在蛋白质界面。为此,我们从欧洲生物信息学研究所(AF-EBI)数据库19,41中获得了人类蛋白质的所有结构和模型。该数据库目前包括高度精确的结构,许多实际上包含具有实验解决的结构的结构域,在PDB中没有结构或与PDB结构几乎没有同源性但通过AlphaFold预测局部距离差异测试(pLDDT)和预测比对误差(PAE)判断高度精确的模型,以及几个非常低的pLDDT和PAE分数的模型。我们根据pLDDT和PAE得分从总共20504个条目中选择了7464个高质量模型进行进一步分析,如方法中所述。
我们可以立即注意到,我们的模型产生了稳健的结果,进一步验证了界面预测的质量。特别地,特定分子界面的氨基酸分布概括了已知的生物化学(例如,Arg和Lys残基主要参与核酸相互作用,疏水氨基酸参与脂质结合位点等)。参见补充图7、8)。此外,将预测的接口映射到UniProt注释的特征显示了与绑定接口的预期功能角色的强烈一致(图4a和补充数据1)。对预测质量的额外支持来自预测界面及其亚细胞定位、GO功能和过程的绘图(补充图9-19)。
我们进一步询问了人类界面组的预测界面的几何特征,并观察到当计算它们的溶剂可及表面积(SASA)时,与蛋白质和核酸的相互作用分别涉及32±22和29±23 nm2的最大面积,而配体和离子涉及16±7和7±4 nm2的小区域。蛋白质-脂质相互作用的SASA分布具有双峰分布,反映了特定的脂质结合位点(17±9 nm2)和跨膜蛋白域周围的大脂质冠状物(75±19 nm2,补充图20)。
作为进一步的验证,将分析扩展到另一个真核生物蛋白质组,我们将PeSTo预测与AlphaFold和RoseTTAFold42衍生的酵母蛋白质组的蛋白质二元复合物的可用预测进行了比较。同样在这种情况下,我们观察到界面中涉及的残基组之间具有非常好的相关性,ROC AUC稳步增加,因为分析限于更高质量模型的区域(图4d)。此外,我们鉴定了额外的结合界面,其可以进一步扩展二元复合物的相互作用网络,并可以用作补充手段来更好地描述和模拟大蛋白质复合物的结构(补充图21)。
值得注意的是,突变位点的47%的UniProt注释落在预测的界面中,28%对应于致病性天然变异位点,14%对应于良性天然变异位点,随机残基的基线为19%在界面内(图4b)。由于我们在PeSTo网站上完全提供了所有这些预测,并且在EBI数据库中可以免费获得潜在的结构模型,因此细胞生物学家可以直接咨询这些致病突变的确切位置以及它们可能损害的相互作用,以便开发合理的工作假说,从而帮助进一步的治疗开发。
继续对预测界面进行大规模分析,我们观察到某些种类界面的强烈分离和其他界面的相当大的重叠(图4c和补充图22)。前一种情况的一个例子是蛋白质界面倾向于与高度分离的蛋白质或离子/配体相互作用。进一步研究这些模式可能有助于发现变构调节机制。在以相当广泛的重叠为特征的成对界面中,那些介导与其他蛋白质和脂质相互作用的界面可能指向膜上可逆的蛋白质二聚化/寡聚化。在实际应用PeSTo解决生物学问题时,应仔细研究具体案例,重叠或缺乏重叠可能会带来如下举例说明的信息。
重要的是,人类蛋白质组以及其他蛋白质组的高分辨率结构和高质量AlphaFold模型的可用性,为生物学家提供了一个机会,可以立即轻松地询问他们感兴趣的蛋白质的特定相互作用预测,快速开发有效的假设,并设计新的实验,从而发现新的生物学。
预测形成蛋白质结合界面的另一组残基定位于跨膜区外的4个位置(图4e)。在细胞膜的胞质侧,三个具有强预测蛋白相互作用潜力的STRA6片段定位于由两个折叠元件组成的位点,这两个折叠元件与Berry等人43实际上提出的序列片段重叠,作为调节细胞视黄醇结合蛋白1 (CRBP1)的结合位点,与对应于已知激酶结合位点(JAK2)的预测相互作用位点相邻。在膜的细胞外侧,还预测了载体视黄醇结合蛋白(RBP)的预期结合位点。因此,具有高蛋白质相互作用分数的残基(例如,报道的RBP的K324-K348,报道的CRBP1位点周围的L251R257和R638-L46,以及激酶位点的D612-K626,图4e)是旨在探测各种相互作用的诱变研究的潜在候选物。
第二个值得描述的例子是PRAMEfm1,在UniProt中被注释为可能通过转录的负调节与细胞分化、增殖和凋亡过程相关。这种蛋白质与一些核糖核酸酶抑制因子具有非常弱的序列同源性,并且高置信度α折叠模型发现除了N-末端部分的某些插入和变形之外,与其中一些具有实质上的结构相似性。
与PeSTo对核糖核酸酶抑制因子的预测相反,其完全局限于已知的核糖核酸酶结合界面(补充图24),在PRAMEfm1的AlphaFold-EBI模型上,PeSTo检测到两个易于蛋白质相互作用的清晰区域。C-末端一半的区域可以容纳蛋白质,类似于核糖核酸酶抑制剂如何与核糖核酸酶相互作用,这立即表明一组残基的突变将破坏与该侧蛋白质靶标的相互作用(例如,由H243、T278、G303、Q360、N387、L422、T455和C-末端P464-L472周围的β-转角的短片段组成的rim)。
PeSTo预测的第二个蛋白质界面位于N-末端的一半,它包括一个低置信度的短片段,可能是无序的,并与PeSTo预测的结合核酸的大表面区域重叠。这是另一个明确的区域,其作用可以通过靶向残基L122Q145以及可能的连接β链进行实验研究。虽然很难从这种计算分析中得出PRAMEfm1的具体作用,但在UniProt注释的背景下,PeSTo预测将提示其作为连接其他蛋白质(通过C-末端部分)和核酸(通过N-末端部分)的枢纽的作用,鉴于其细胞质定位,可能是RNA,并可能受也结合到N-末端部分的其他蛋白质的调节。
我们最后比较了PeSTo的蛋白质-蛋白质界面预测和使用AlphaFoldmultimer16模拟蛋白质-蛋白质相互作用,这是一种信息更丰富的方法,也包括进化偶联。在STRA6的例子中,α折叠多聚体预测CRBP1在我们从文献中讨论的相同残基周围结合到STRA6上,即基本上与PeSTo相同的预测。
然而,α折叠多聚体根本不能预测JAK2的任何相互作用,并预测RBP的不正确结合位点。在PRAMEfm1的情况下,我们检测到一个似是而非的核酸结合界面,这是AlphaFold没有训练预测的,我们检测到一个高度可信的蛋白质相互作用区域,但没有任何关于配偶体身份的信息,排除了用AlphaFold测试任何明显的、特异性的复合物。这些比较突出了PeSTo和α折叠多聚体在预测蛋白质-蛋白质相互作用方面的协同交叉。也就是说,PeSTo可以产生与报道的生物化学一致的预测,而当相互作用网络已知时,α折叠多聚体可以询问这些结合界面。
与现有方法比较
我们的方法与ScanNet15、MaSIF-site7、36、SPPIDER35和PSIVER37进行了比较。ScanNet是最新的基于几何的深度学习方法,用于蛋白质-蛋白质界面预测。MaSIF-site是蛋白质-蛋白质界面预测的最佳可用的基于表面的深度学习方法。SPPIDER是一种历史悠久、久经考验的方法,用作蛋白质-蛋白质界面预测的参考。PSIVER只使用序列信息,并进行基准测试,以显示基于结构的方法和基于序列的方法之间的性能差异。PeSTo的基准测试是使用专门取自测试数据集的结构进行的。对于蛋白质、离子和配体界面的预测,我们使用每种界面类型512种结构。对于核酸和脂质界面预测,可用结构的低数量将测试数据集分别限制为391和161个结构。
讨论
我们在这里表明,蛋白质原子坐标的几何变换足以以高分辨率检测和分类蛋白质结合界面,超过了其他方法的预测能力,而不需要明确描述系统的物理和化学性质,因此没有预先计算分子表面和/或附加性质的开销。所有这些使用适度的计算资源并以非常高的速度实现了对大型结构系综的分析,例如由分子动力学模拟产生的那些,这揭示了研究蛋白质相互作用网络的动态特征的机会。
同样,大型结构数据集,如由最新一代三级蛋白质结构预测工具创建的数据集,可以很容易地进行分析,就像这里对人类基因组所做的那样,并有可能快速获得新的生物学发现。
为了使基于PeSTo的蛋白质预测对社区可用,我们在https://pesto.epfl.ch/,的一个web服务器上实现了它,无需注册即可免费访问。服务器采用PDB格式的任何蛋白质结构和模型(上传或从PDB或阿尔法折叠-EBI数据库中获取),并返回它们和基于每个残基的预测置信度的附加信息报告。输出文件可以下载或可视化的权利在网站上。此外,我们还提供了源代码(https://github.com/LBM-EPFL/PeSTo ),以便于应用到大型结构集合中,就像这里为人机界面me所做的那样。
如果有足够的训练数据可用,该方法可以很容易地升级(例如改进进一步的蛋白质-脂质预测),并可重复用于其他特定应用。事实上,无参数的PeSTo架构是足够通用的,可以很容易地适应其他基于结构的问题,如对接或建模与材料的相互作用。该描述完全不知道结构中原子的确切物理化学性质,因此容易扩展到其他材料和领域,并且与需要表面和体积的中间计算的方法相比,可能对与起始结构相关的问题(例如缺少原子)不太敏感。
鉴于结构信息的不断积累和预测折叠体数据的快速膨胀,PeSTo是一种准确、灵活、快速和用户友好的解决方案,可用于剖析蛋白质的巨大和动态相互作用景观,并可用于发现新的和更丰富的生物学见解。