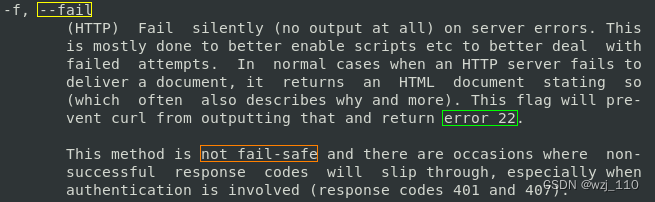
今天给同学们分享一篇生信文章“Identification of new co-diagnostic genes for sepsis and metabolic syndrome using single-cell data analysis and machine learning algorithms”,这篇文章发表Front Genet.期刊上,影响因子为3.7。

结果解读:
常见差异基因的筛选
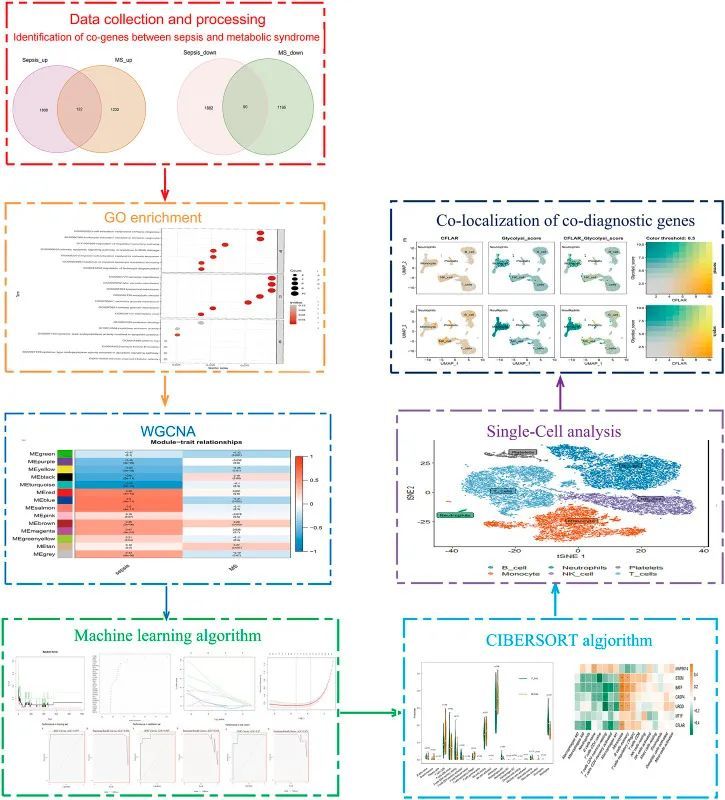
如图1所示,研究流程图解释了它是如何进行的。在校正和归一化之前,在三个数据集(GSE28750、GSE154918和GSE98895)上进行PCA。数据集被标准化,在脓毒症中发现3902个DEG(1930个上调和1972个下调),而在MetS中发现2639个DEGs(1354个上调和1285个下调)。通过鉴定脓毒症和代谢综合征之间的常见DEG,发现了122个常见的上调DEG和90个常见的下调DEG(图2A、B)。对已鉴定的常见DEG进行GO富集分析,以研究其生物学功能和途径。根据GO分析,常见上调的DEG主要参与细胞活化和白细胞活化,参与免疫反应和调节分泌途径,而常见下调的DEG富集于上皮
WGCNA共表达基因模块的分析
在阈值为80的情况下,检测并去除了2个异常样本,保留了98个样本。“WGCNA”软件包的“pick Soft Threshold”功能用于过滤1到30之间的功率参数。作为软阈值,选择6的幂以确保无标度网络(图3A)。使用“叉树”动态和模块特征基因函数,共获得了14个包含具有相似共表达性状的基因的模块(图3B)。热图显示了每个模块与疾病之间的相关性(图3C)。“棕色”模块表明脓毒症和代谢综合征高度相关(脓毒症:r=0.46,p=0.009;代谢综合征:r=0.26,p=0.003)。脓毒症与代谢综合征在棕色模块中具有正相关基因(脓毒病:cor=0.38,p=2.8e-18;代谢综合症:cor=0.37,p=2.4e-17)(图3D,E)。对该棕色模块基因进行GO分析。
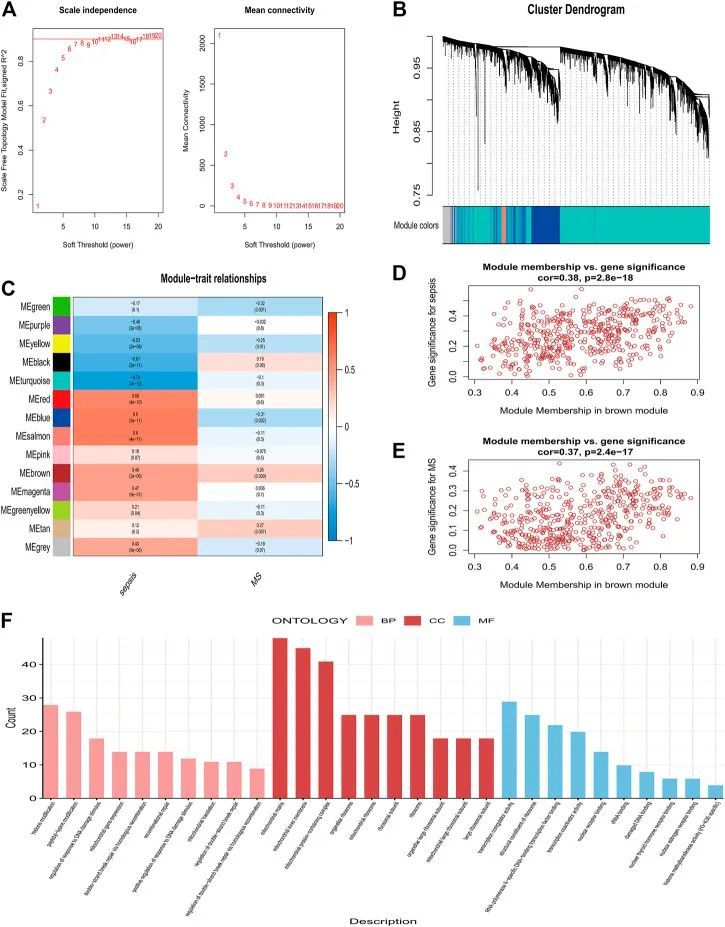
利用机器学习识别候选关键基因
作者使用RF算法结合LASSO回归,最终获得了7个诊断基因,包括STOM、BATF、CASP4、MAP3K14、MT1F、CFLAR、UROD(图4A–D)。之后,作者评估了这些基因的诊断价值。ROC曲线的AUC值分别为STOM的0.995、BATF的0.996、CASP4的0.995、MAP3K14的0.995。所有7个基因特征的AUC>0.9具有较高的准确性,证明了它们的预测能力。基于训练集GSE154918,作者构建了候选基因模型(STOM、BATF、CASP4、MAP3K14、MT1F、CFLAR),并在验证集GSE28750上对其进行了评估。如图4E所示,在GSE154918中,ROC值的AUC为0.997,PR值为0.995。
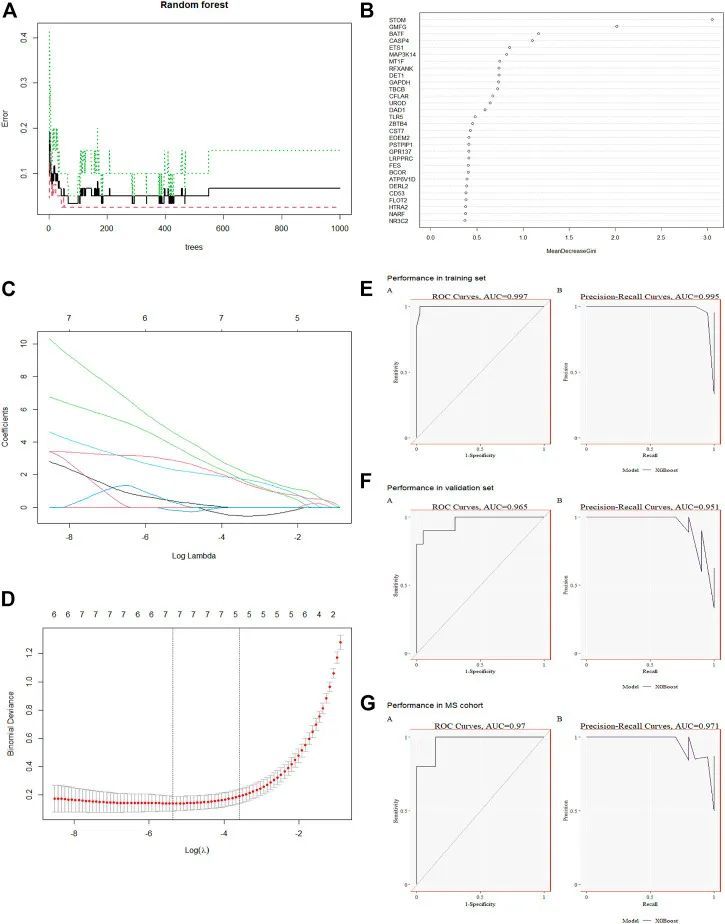
脓毒症和代谢综合征患者免疫细胞的浸润
对有免疫浸润的脓毒症和代谢综合征患者进行了研究。此外,热图显示了免疫细胞中七个关键基因的差异表达(图5B,D)。正常组织比脓毒症组织含有更少的中性粒细胞和单核细胞(p<0.05)。脓毒症患者组织和正常组织的比较显示,脓毒症的组织含有明显更少的幼稚B细胞、记忆幼稚B细胞,CD8幼稚T细胞和CD4幼稚T细胞(图5A)。STOM、BATF、CASP4、MT1F、CFLAR和UROD的表达与静息NK细胞、CD4幼稚T细胞、CD8 T细胞和CD4静息T细胞的浸润水平呈负相关。MAP3K14的表达与中性粒细胞、活化肥大细胞、单核细胞、巨噬细胞M0和NK活化细胞呈负相关(图5B)。
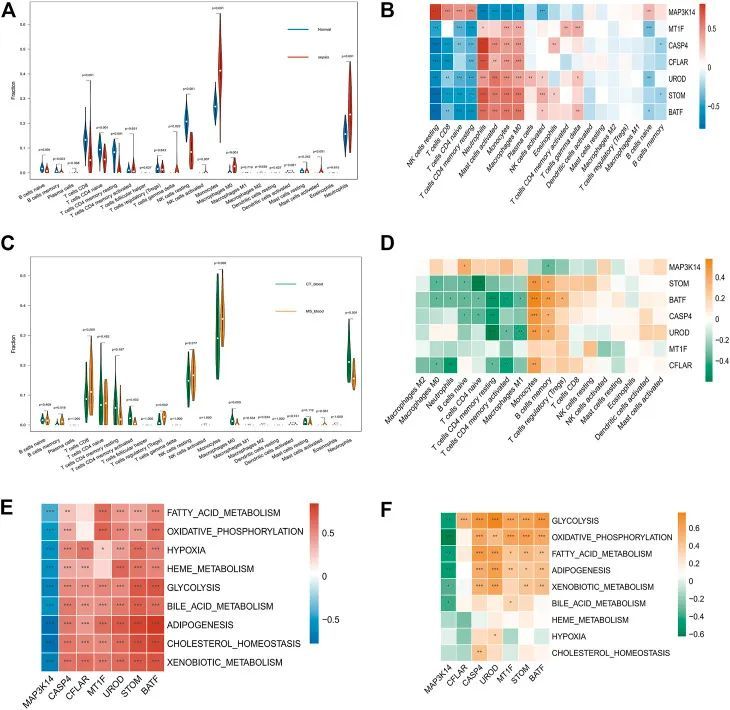
脓毒症和正常患者的单细胞测序分析
为了检查单细胞数据集GSE167363的质量,进行了初步质量检查。nFeature RNA、nCount RNA和prent之间的相关性。对mt进行了检测,以确保研究中使用的细胞样品具有高质量。图6A显示了nCount RNA和代表独特分子标识符的nFeature RNA之间的正相关,相关系数为0.94。作者排除了一些细胞,结果如图6B、C所示。在scRNA-seq数据集中,共鉴定了3000个具有高水平变异的基因,并标记了10个最重要的标记。对前20个PC进行了主成分分析(图6D)。使用t-SNE算法对细胞进行聚类,获得21个聚类(图7A)。
总结
使用单细胞分析和WGCNA以及机器学习技术的组合来鉴定脓毒症和MetS中涉及的效应基因。此外,还发现疾病诊断基因与多种免疫细胞和代谢途径有关。葡萄糖代谢相关途径可能在脓毒症和代谢综合征中都很常见,在脓毒症患者中,葡萄糖代谢可能通过单核细胞和NK细胞发挥作用。作者发现CFLAR基因可能在脓毒症患者的葡萄糖代谢中发挥关键作用。本研究可能为脓毒症的诊断和治疗提供一种新的方法。