目录
GSVA
1:获取注释基因集
2:运行
GSEA
1,示例数据集
2,运行
GSEA_KEGG富集分析
GSEA_GO富集分析
DO数据库GSEA
MSigDB数据库选取GSEA
KEGG
1:运行
2:绘图
bar图
气泡图
绘图美化
GO
GSVA
1:获取注释基因集
2:运行
GSEA
1,示例数据集
2,运行
GSEA_KEGG富集分析
GSEA_GO富集分析
DO数据库GSEA
MSigDB数据库选取GSEA
KEGG
GO
GSVA
【精选】RNA 18. SCI 文章中基因集变异分析 GSVA_gsva分析-CSDN博客
RNA-seq入门实战(八):GSVA——基因集变异分析 - 知乎 (zhihu.com)
表达矩阵反映了样本和基因的关系,则GSVA将一个“样本×基因”的矩阵转化为“样本×通路”的矩阵,直接反映了样本和读者感兴趣的通路之间的联系。因此,如果用limma包做差异表达分析可以寻找样本间差异表达的基因,同样地,使用limma包对GSVA的结果(依然是一个矩阵)做同样的分析,则可以寻找样本间有显著差异的通路。这些“差异表达”的通路,相对于基因而言,更加具有生物学意义,更具有可解释性,是统计学与生物学成功结合后,对GSEA结果的一次升华,可以进一步用于肿瘤subtype的分型等等与生物学意义结合密切的探究。
1:获取注释基因集
可以用msigdbr包下载读取或者GSEA | MSigDB | Human MSigDB Collections (gsea-msigdb.org)下载整理。 按需下载整理

##进行数据读取
geneSets <- getGmt('c2.cp.kegg_medicus.v2023.2.Hs.symbols.gmt') ###下载的基因集##下载symbols的 注释文件,使用表达矩阵是省略了转换的麻烦2:运行
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)library(GSVA)
library(GSEABase)
library(clusterProfiler)expr <- read.csv("easy_input_expr.csv", row.names = 1)#表达矩阵
geneSets <- getGmt('h.all.v2023.2.Hs.symbols.gmt')##symbols 较为方便##运行
GSVA_hall <- gsva(expr=as.matrix(expr),#需要为matrix格式gset.idx.list=geneSets, # method="gsva", #c("gsva", "ssgsea", "zscore", "plage")mx.diff=T, # 数据为正态分布则T,双峰则Fkcdf="Gaussian", #CPM, RPKM, TPM数据就用默认值"Gaussian", read count数据则为"Poisson",parallel.sz=4,# 并行线程数目min.sz=2) GSEA
快速拿捏KEGG/GO/Reactome/Do/MSigDB的GSEA富集分析! (qq.com)
【精选】RNA 11. SCI 文章中基因表达富集之 GSEA_gsea数据库-CSDN博客
GSEA(Gene Set EnrichmentAnalysis),即基因集富集分析,它的基本思想是使用预定义的基因,将基因按照在两类样本中的差异表达程度排序,然后检验预先设定的基因集合是否在这个排序表的顶端或者底端富集。
1,示例数据集
rm(list = ls())
library(DOSE)##包里有测试数据
library(clusterProfiler)
require(enrichplot)
options(stringsAsFactors = F)data(geneList, package = "DOSE")
head(geneList)##DOSE提供的一个geneList,name是每一个entrez gene id, value是log2FoldChange值。
df <- as.data.frame(geneList)##查看:还原gene symbol
df$ENTREZID <- rownames(df)
#?bitr 函数查看
df1<-bitr(df$ENTREZID, #转换的列是df数据框中的SYMBOL列fromType = "ENTREZID",#需要转换ID类型toType = "SYMBOL",#转换成的ID类型OrgDb = "org.Hs.eg.db")#物种选择(小鼠的是org.Mm.eg.db)
df2<-merge(df,df1,by="ENTREZID",all=F)##进行合并
测试数据查看

2,运行
GSEA_KEGG富集分析
rm(list = ls())
library(DOSE)##包里有测试数据
library(clusterProfiler)
require(enrichplot)
options(stringsAsFactors = F)
##input需要的是entrez ID+log2fc文件(包里的文件即为entrez ID+log2fc)
##如果不是entrez ID+log2fc,需要进行转换##数据集查看和整理
data(geneList, package = "DOSE")
head(geneList)
#4312 8318 10874 55143 55388 991
#4.572613 4.514594 4.418218 4.144075 3.876258 3.677857
df <- as.data.frame(geneList)##查看:还原gene symbol
df$ENTREZID <- rownames(df)
#?bitr 函数查看
df1<-bitr(df$ENTREZID, #转换的列是df数据框中的SYMBOL列fromType = "ENTREZID",#需要转换ID类型toType = "SYMBOL",#转换成的ID类型OrgDb = "org.Hs.eg.db")#物种选择(小鼠的是org.Mm.eg.db)
df2<-merge(df,df1,by="ENTREZID",all=F)##进行合并
df3 <-df2[,-1]
head(df3)#自己分析常见的格式
#geneList SYMBOL
#1 -0.28492113 NAT2##以df3 为input进行转换:添加entrez ID列,symbol转entrez ID
##注意还原结果少几十个基因:因为一个entrez ID可能对应多个symbol(一基因多symbol)
dat<-bitr(df3$SYMBOL, fromType = "SYMBOL", #现有的ID类型toType = "ENTREZID",#需转换的ID类型OrgDb = "org.Hs.eg.db")#物种
head(dat)##转换时部分SYMBOL会转换失败dat1<-merge(df3,dat,by="SYMBOL",all=F)##进行合并
#按照foldchange排序
sortdf<-dat1[order(dat1$geneList, decreasing = T),]#这里geneList其实是logFC值
head(sortdf)geneList1 <- sortdf$geneList##先把foldchange按照从大到小提取出来
names(geneList1) <- sortdf$ENTREZID###给上面提取的foldchange加上对应上ENTREZID
head(geneList1 )
#4312 8318 10874 55143 55388 991
#4.572613 4.514594 4.418218 4.144075 3.876258 3.677857 #GSEA_KEGG富集分析:
KEGG_ges <- gseKEGG(geneList = geneList1,#inputorganism = "hsa")#物种#按照enrichment score从高到低排序,便于查看富集通路
sortKEGG_ges<-KEGG_ges[order(KEGG_ges$enrichmentScore, decreasing = T),]
#sortKEGG_ges <- KEGG_ges@result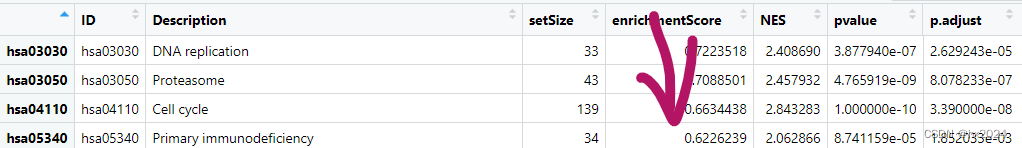
说明:在GSEA中,基因集的富集分数可以为正或负,表示该基因集在对应生物条件下的富集程度。富集分数的绝对值越大,表示富集程度越高。 对于AB两个亚型的差异基因,在GSEA富集分析中,结果按照富集分数排序。通常情况下,富集分数为正最大的前几个表示在A亚型中富集的集中的通路,而富集分数负值最大的几个表示在B亚型中富集的集中的通路。 需要注意的是,富集分数的正负并不代表富集的方向,而是表示富集程度的大小。因此,正值最大的前几个通路表示在A亚型中富集程度最高的通路,负值最大的几个通路表示在B亚型中富集程度最高的通路。 这样的排序方式可以帮助我们理解不同亚型或条件下基因集的富集模式,以及与这些通路相关的生物学过程和功能。
#进行绘图
gseaplot2(KEGG_ges, row.names(sortKEGG_ges)[1:5])##可以自行选择通路
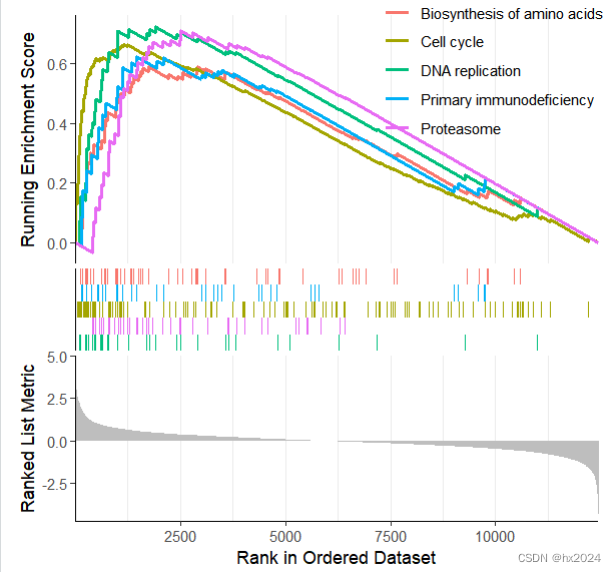
dev.off()
#个性化展示(选取结果中的ID)
p1 <- gseaplot2(KEGG_ges,geneSetID = c("hsa03030","hsa03050","hsa04710","hsa00350"),#通路color = c("#003399", "#FFFF00", "#FF6600","black"),#颜色pvalue_table = TRUE,#显示P值ES_geom = "line")#"dot"将线转换为点
p1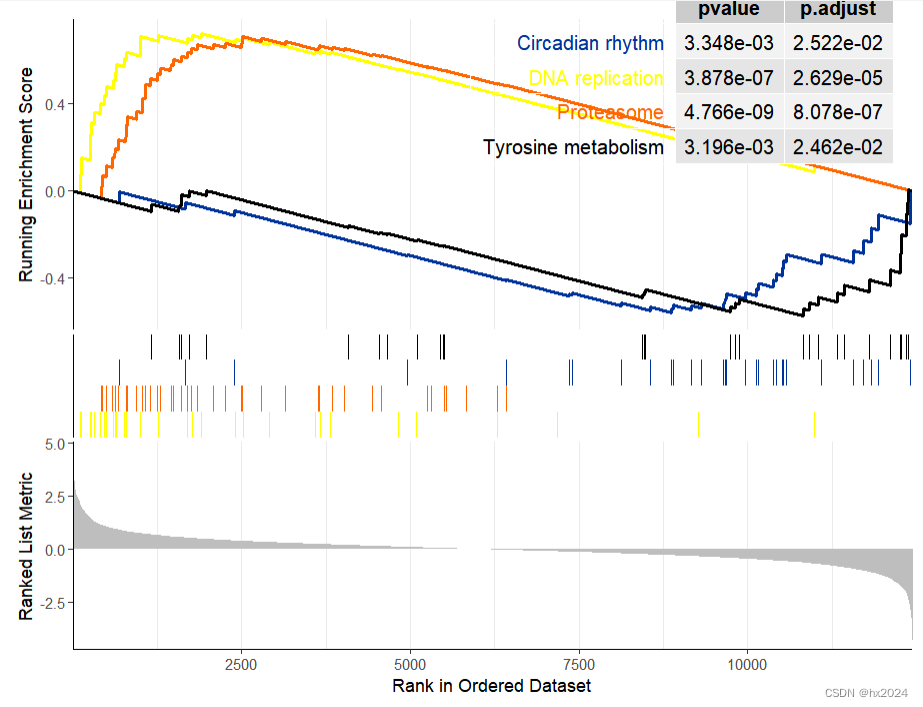
GSEA_GO富集分析
主要是函数的不同 gseGO 函数
#GSEA
rm(list = ls())
library(DOSE)##包里有测试数据
library(clusterProfiler)
require(enrichplot)
options(stringsAsFactors = F)##数据集查看和整理
data(geneList, package = "DOSE")
head(geneList)##GSEA_GO富集分析:
GO_ges <- gseGO(geneList = geneList,OrgDb = "org.Hs.eg.db",ont = "CC", #one of "BP", "MF", and "CC" subontologies, or "ALL" for all three.minGSSize = 10,maxGSSize = 500,pvalueCutoff = 0.05,eps = 0,verbose = FALSE)
#res <- GO_ges@result
res1<-GO_ges[order(GO_ges$enrichmentScore, decreasing = T),]DO数据库GSEA
需要library(DOSE) DO(Disease Ontology)数据库GSEA
#GSEA
rm(list = ls())
library(DOSE)##包里有测试数据
library(clusterProfiler)
require(enrichplot)
options(stringsAsFactors = F)##数据集查看和整理
data(geneList, package = "DOSE")
head(geneList)##GSEA_DO(Disease Ontology)富集分析:
DO_ges <- gseDO(geneList,minGSSize = 10,maxGSSize = 500,pvalueCutoff = 0.05,pAdjustMethod = "BH",verbose = FALSE,eps = 0)#res <- DO_ges@result
res1<-DO_ges[order(DO_ges$enrichmentScore, decreasing = T),]MSigDB数据库选取GSEA
msigdf + clusterProfiler全方位支持MSigDb (guangchuangyu.github.io)
Q&A | 如何使用clusterProfiler对MSigDB数据库进行富集分析 - 知乎 (zhihu.com)
#GSEA
rm(list = ls())
library(DOSE)##包里有测试数据
library(clusterProfiler)
require(enrichplot)
library(msigdbr)
options(stringsAsFactors = F)##数据集查看和整理
data(geneList, package = "DOSE")
head(geneList)##msigdbr 提取注释自己所需的注释基因集
H <- msigdbr(species = "Homo sapiens", category = "H") %>% dplyr::select(gs_name, entrez_gene)#C2all <- msigdbr(species = "Homo sapiens",
# category = "H")#完整的注释##富集分析
H_ges <- GSEA(geneList,TERM2GENE = H,##注释基因集minGSSize = 10,maxGSSize = 500,pvalueCutoff = 0.05,pAdjustMethod = "BH",verbose = FALSE,eps = 0)#res <- H_ges@result
res1<-H_ges[order(H_ges$enrichmentScore, decreasing = T),]KEGG
KEGG富集分析及可视化,一把子拿捏! (qq.com)
RNA 10. SCI 文章中基因表达富集之 KEGG 注释_kegg中qvalue_桓峰基因的博客-CSDN博客
在线KEGGDAVID Functional Annotation Bioinformatics Microarray Analysis (ncifcrf.gov)
DAVID 在线数据库进行 GO/ KEGG 富集分析_david数据库go富集分析-CSDN博客
1:运行
##差异基因KEGG富集分析
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(dplyr)#数据清洗
library(org.Hs.eg.db)#ID转换
library(clusterProfiler)#富集分析
library(ggplot2)#绘图
library(RColorBrewer)#配色调整
library(DOSE)##包里有测试数据data(geneList, package = "DOSE")
head(geneList)
df <- as.data.frame(geneList)##查看:还原gene symbol
df$ENTREZID <- rownames(df)
#?bitr 函数查看
df1<-bitr(df$ENTREZID, #转换的列是df数据框中的SYMBOL列fromType = "ENTREZID",#需要转换ID类型toType = "SYMBOL",#转换成的ID类型OrgDb = "org.Hs.eg.db")#物种选择(小鼠的是org.Mm.eg.db)
df2<-merge(df,df1,by="ENTREZID",all=F)##进行合并##选择logFC>1.5的基因:我们以这个筛选的差异基因集为input测试
df3 <- df2[abs(df2$geneList)>1.5,]##abs() 表示绝对值
##自己使用时进行自定义KEGG_diff <- enrichKEGG(gene = df3$ENTREZID,organism = "hsa",#物种,Homo sapiens (human)pvalueCutoff = 0.05,qvalueCutoff = 0.05)KEGG_result <- KEGG_diff@result#保存富集结果:
save(KEGG_diff,KEGG_result,file = c("KEGG_diff.Rdata"))
#write.csv(KEGG_result,file = "KEGG_result.csv")
ID:pathway的ID名;GeneRatio:差异基因中富集到该pathway的基因数目/富集到所有pathway的总差异基因数目;BgRatio:所有背景基因中富集到该pathway的基因数目/总背景基因数目;
Count:富集到该pathway的基因数目
2:绘图
bar图
#绘图
library(enrichplot)
library(stringr)
library(cowplot)
library(ggplot2)
barplot(KEGG_diff,x = "Count", #or "GeneRatio"color = "pvalue", #or "p.adjust" and "qvalue"showCategory = 20,#显示前top20font.size = 12,title = "KEGG enrichment barplot",label_format = 30 #超过30个字符串换行
)
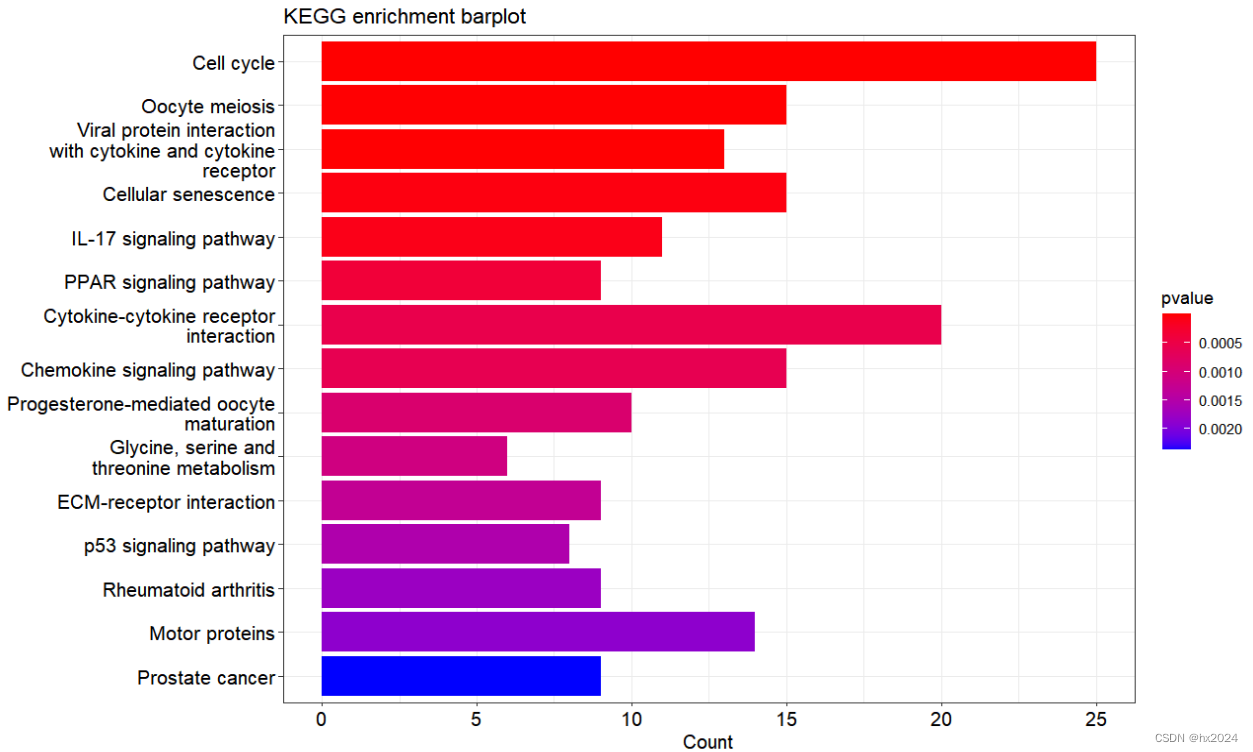
气泡图
dotplot(KEGG_diff,x = "GeneRatio",color = "p.adjust",title = "Top 20 of Pathway Enrichment",showCategory = 20,label_format = 30
)
绘图美化
#将pathway按照p值排列
KEGG_top20 <- KEGG_result[1:20,]
KEGG_top20$pathway <- factor(KEGG_top20$Description,levels = rev(KEGG_top20$Description))
p2 <- ggplot(data = KEGG_top20,aes(x = Count,y = pathway))+geom_point(aes(size = Count,color = -log10(pvalue)))+ # 气泡大小及颜色设置theme_bw()+scale_color_distiller(palette = "Spectral",direction = 1) +labs(x = "Gene Number",y = "",title = "Dotplot of Enriched KEGG Pathways",size = "Count")
p2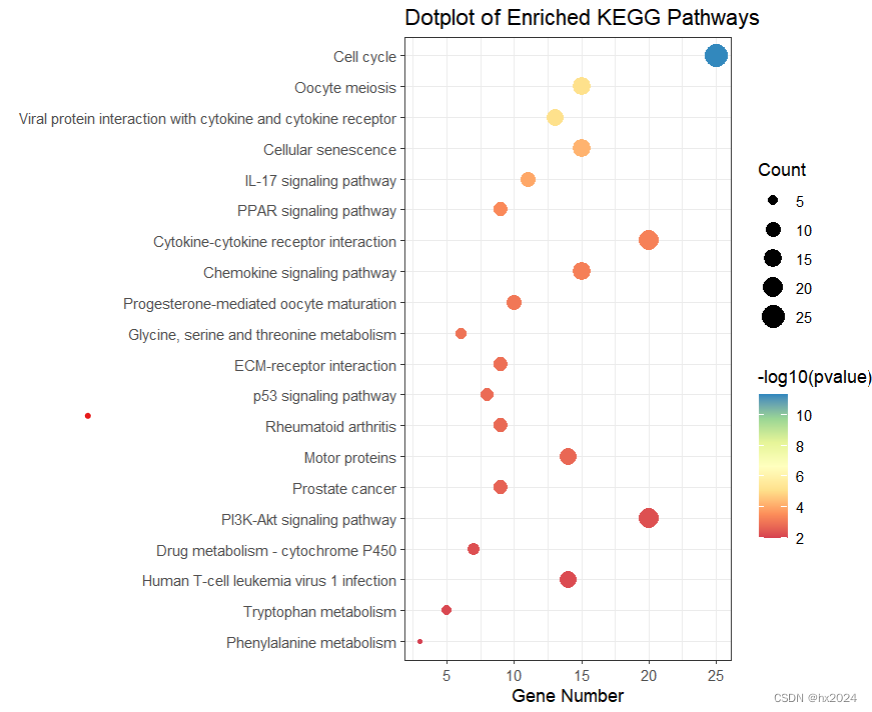
GO
GO富集分析及可视化,一把子拿捏! (qq.com)
【精选】RNA 9. SCI 文章中基因表达之 GO 注释_consider increasing max.overlaps_桓峰基因的博客-CSDN博客
就是函数的差别
GO_MF_diff <- enrichGO(gene = diff_entrez$ENTREZID, #用来富集的差异基因
OrgDb = org.Hs.eg.db, #指定包含该物种注释信息的org包
ont = "MF", #可以三选一分别富集,或者"ALL"合并
pAdjustMethod = "BH", #多重假设检验矫正方法
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE) #是否将gene ID映射到gene name
#提取结果表格:
GO_MF_result <- GO_MF_diff@result
View(GO_MF_result)感谢上面的许多教程:更详细的大家可以去学习!