介绍:
GitHub - EBI-Metagenomics/EukCC: Tool to estimate genome quality of microbial eukaryotes
安装:
docker:
docker pull microbiomeinformatics/eukcc推荐conda 环境:
conda install -c conda-forge -c bioconda "eukcc>=2"
# mamba更快
mamba install -c conda-forge -c bioconda "eukcc>=2"pip install eukcc数据库配置,docker记得映射目录
mkdir eukccdb
cd eukccdb
wget http://ftp.ebi.ac.uk/pub/databases/metagenomics/eukcc/eukcc2_db_ver_1.1.tar.gz
tar -xzvf eukcc2_db_ver_1.1.tar.gz数据库下载地址:Index of /pub/databases/metagenomics/eukcc
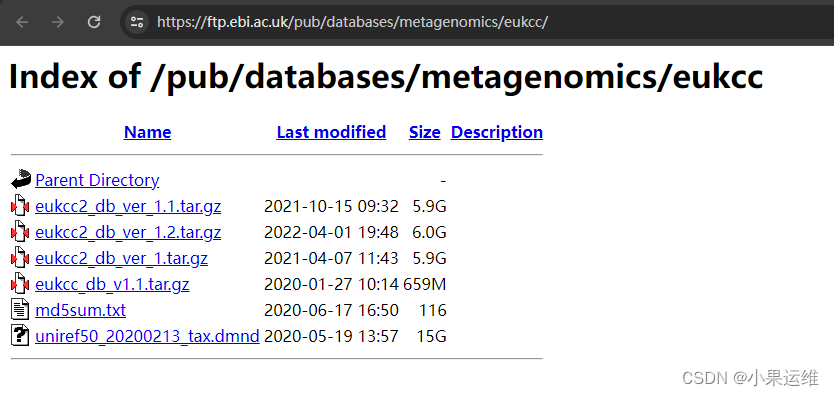
下载数据库注意版本,一般选版本2吧,
链接:https://ftp.ebi.ac.uk/pub/databases/metagenomics/eukcc/eukcc2_db_ver_1.2.tar.gz
https://ftp.ebi.ac.uk/pub/databases/metagenomics/eukcc/eukcc_db_v1.1.tar.gz
还有个副产品,diamond的数据库,不过好像看不出是diamond的哪个版本生成的,用的时候不好用的话就用下载的数据库再生成一遍吧。
https://ftp.ebi.ac.uk/pub/databases/metagenomics/eukcc/uniref50_20200213_tax.dmnd
如果不知道数据库位置,或者软件找不到位置,那就简单吧,设置DB目录
export EUKCC2_DB=/path/to/.../eukcc2_db_ver_1.1
快速开始
#EukCC on a single MAG
#We assume that you did set you $EUKCC2_DB to the correct location. If not please use the --db flag to pass the database to EukCC.eukcc single --out outfolder --threads 8 bin.fa
#EukCC will then run on 8 threads. You can pass nucleotide fastas or proteomes to EukCC. It will automatically try to detect if it has to predict proteins or not.#By default it will never use more than a single threads for placing the genomes in the reference tree, to save memory.#EukCC on a folder of bins
eukcc folder --out outfolder --threads 8 bins
#EukCC will assume that the folder contains files with the suffix .fa. If that is not the case please adjust the parameter.序列拼接流程
双端序列需要先构建bam索引
cat binfolder/*.fa > pseudo_contigs.fasta
bwa index pseudo_contigs.fasta
bwa mem -t 8 pseudo_contigs.fasta reads_1.fastq.gz reads_2.fastq.gz |samtools view -q 20 -Sb - |samtools sort -@ 8 -O bam - -o alignment.bam
samtools index alignment.bam利用py脚本生成关联表
binlinks.py --ANI 99 --within 1500 \--out linktable.csv binfolder alignment.bamIf you have multiple bam files, pass all of them to the script (e.g. *.bam).
You will obtain a three column file (bin_1,bin_2,links).
拼接bins
eukcc folder \--out outfolder \--threads 8 \--links linktable.csv \binfolderEukCC 首先将分别对所有bins进行运行。随后,它会识别那些至少达到50%完整度但尚未超过100-improve_percent的中等质量bins。接下来,它会找出那些通过至少100对端读配对与这些中等质量bins相连接的bins。若经过合并后bin的质量评分有所提高,则该bin将会被合并。
已合并的bins可以在输出文件夹中找到。
警示
blinks.py
#!/usr/bin/env python3
import pysam
from Bio import SeqIO
from collections import defaultdict
import os
import argparse
import logging
import csvdef is_in(read, contig_map, within=1000):if read.reference_name not in contig_map.keys():return Falseif read.reference_start <= within or read.reference_end <= within:return Trueelif read.reference_start > (contig_map[read.reference_name] - within) or read.reference_end > (contig_map[read.reference_name] - within):return Trueelse:return Falsedef keep_read(read, contig_map, within=1000, min_ANI=98, min_cov=0):ani = ((read.query_alignment_length - read.get_tag("NM"))/ float(read.query_alignment_length)* 100)cov = read.query_alignment_length / float(read.query_length) * 100if ani >= min_ANI and cov >= min_cov and is_in(read, contig_map, within) is True:return Trueelse:return Falsedef contig_map(bindir, suffix=".fa"):m = {}for f in os.listdir(bindir):if f.endswith(suffix) is False:continuepath = os.path.join(bindir, f)with open(path, "r") as handle:for record in SeqIO.parse(handle, "fasta"):m[record.name] = len(record.seq)return mdef bin_map(bindir, suffix=".fa"):contigs = defaultdict(str)contigs_per_bin = defaultdict(int)for f in os.listdir(bindir):if f.endswith(suffix) is False:continuepath = os.path.join(bindir, f)binname = os.path.basename(f)with open(path, "r") as handle:for record in SeqIO.parse(handle, "fasta"):contigs[record.name] = binnamecontigs_per_bin[binname] += 1return contigs, contigs_per_bindef read_pair_generator(bam):"""Generate read pairs in a BAM file or within a region string.Reads are added to read_dict until a pair is found.From: https://www.biostars.org/p/306041/"""read_dict = defaultdict(lambda: [None, None])for read in bam.fetch():if not read.is_paired or read.is_secondary or read.is_supplementary:continueqname = read.query_nameif qname not in read_dict:if read.is_read1:read_dict[qname][0] = readelse:read_dict[qname][1] = readelse:if read.is_read1:yield read, read_dict[qname][1]else:yield read_dict[qname][0], readdel read_dict[qname]def read_bam_file(bamf, link_table, cm, within, ANI):samfile = pysam.AlignmentFile(bamf, "rb")# generate link tablelogging.info("Parsing Bam file. This can take a few moments")for read, mate in read_pair_generator(samfile):if keep_read(read, cm, within, min_ANI=ANI) and keep_read(mate, cm, within, min_ANI=ANI):# fill in the tablelink_table[read.reference_name][mate.reference_name] += 1if read.reference_name != mate.reference_name:link_table[mate.reference_name][read.reference_name] += 1return link_tabledef main():# set arguments# arguments are passed to classesparser = argparse.ArgumentParser(description="Evaluate completeness and contamination of a MAG.")parser.add_argument("bindir", type=str, help="Run script on these bins")parser.add_argument("bam",type=str,help="Bam file(s) with reads aligned against all contigs making up the bins",nargs="+",)parser.add_argument("--out","-o",type=str,required=False,help="Path to output table (Default: links.csv)",default="links.csv",)parser.add_argument("--ANI", type=float, required=False, help="ANI of matching read", default=99)parser.add_argument("--within",type=int,required=False,help="Within this many bp we need the read to map",default=1000,)parser.add_argument("--contigs","-c",action="store_true",default=False,help="Instead of bins print contigs",)parser.add_argument("--quiet","-q",dest="quiet",action="store_true",default=False,help="Silcence most output",)parser.add_argument("--debug","-d",action="store_true",default=False,help="Debug and thus ignore safety",)args = parser.parse_args()# define logginglogLevel = logging.INFOif args.quiet:logLevel = logging.WARNINGelif args.debug:logLevel = logging.DEBUGlogging.basicConfig(format="%(asctime)s %(message)s",datefmt="%d-%m-%Y %H:%M:%S: ",level=logLevel,)bindir = args.bindircm = contig_map(bindir)bm, contigs_per_bin = bin_map(bindir)logging.debug("Found {} contigs".format(len(cm)))link_table = defaultdict(lambda: defaultdict(int))bin_table = defaultdict(lambda: defaultdict(int))# iterate all bam filesfor bamf in args.bam:link_table = read_bam_file(bamf, link_table, cm, args.within, args.ANI)logging.debug("Created link table with {} entries".format(len(link_table)))# generate bin tablefor contig_1, dic in link_table.items():for contig_2, links in dic.items():bin_table[bm[contig_1]][bm[contig_2]] += linkslogging.debug("Created bin table with {} entries".format(len(bin_table)))out_data = []logging.debug("Constructing output dict")if args.contigs:for contig_1, linked in link_table.items():for contig_2, links in linked.items():out_data.append({"bin_1": bm[contig_1],"bin_2": bm[contig_2],"contig_1": contig_1,"contig_2": contig_2,"links": links,"bin_1_contigs": contigs_per_bin[bm[contig_1]],"bin_2_contigs": contigs_per_bin[bm[contig_2]],})else:for bin_1, dic in bin_table.items():for bin_2, links in dic.items():out_data.append({"bin_1": bin_1, "bin_2": bin_2, "links": links})logging.debug("Out data has {} rows".format(len(out_data)))# resultslogging.info("Writing output")with open(args.out, "w") as fout:if len(out_data) > 0:cout = csv.DictWriter(fout, fieldnames=list(out_data[0].keys()))cout.writeheader()for row in out_data:cout.writerow(row)else:logging.warning("No rows to write")if __name__ == "__main__":main()scripts/filter_euk_bins.py
#!/usr/bin/env python3
#
# This file is part of the EukCC (https://github.com/openpaul/eukcc).
# Copyright (c) 2019 Paul Saary
#
# This program is free software: you can redistribute it and/or modify
# it under the terms of the GNU General Public License as published by
# the Free Software Foundation, version 3.
#
# This program is distributed in the hope that it will be useful, but
# WITHOUT ANY WARRANTY; without even the implied warranty of
# MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU
# General Public License for more details.
#
# You should have received a copy of the GNU General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
# provides all file operation functions
# used inthis package
import os
import argparse
import subprocess
import logging
import tempfile
import gzip
from multiprocessing import Pool# backup fasta handler, so we can use readonly directories
class fa_class:def __init__(self, seq, name, long_name):self.seq = seqself.name = nameself.long_name = long_namedef __str__(self):return self.seqdef __len__(self):return len(self.seq)def Fasta(path):"""Iterator for fasta files"""entry = Falsewith open(path) as fin:for line in fin:if line.startswith(">"):if entry is not False:entry.seq = "".join(entry.seq)yield entry# define new entrylong_name = line.strip()[1:]name = long_name.split()[0]entry = fa_class([], name, long_name)else:entry.seq.append(line.strip())# yield last oneentry.seq = "".join(entry.seq)yield entrydef gunzip(path, tmp_dir):"""Gunzip a file for EukRep"""if path.endswith(".gz"):fna_path = os.path.join(tmp_dir, "contigs.fna")logging.debug("Going to unzip fasta into {}".format(fna_path))with gzip.open(path, "r") as fin, open(fna_path, "w") as fout:for line in fin:fout.write(line.decode())path = fna_pathlogging.debug("Done unzipping {}".format(fna_path))return pathclass EukRep:"""Class to call and handle EukRep data"""def __init__(self, fasta, eukout, bacout=None, minl=1500, tie="euk"):self.fasta = fastaself.eukout = eukoutself.bacout = bacoutself.minl = minlself.tie = tiedef run(self):# command list will be calledcmd = ["EukRep","--min",str(self.minl),"-i",self.fasta,"--seq_names","-ff","--tie",self.tie,"-o",self.eukout,]if self.bacout is not None:cmd.extend(["--prokarya", self.bacout])subprocess.run(cmd, check=True, shell=False)self.read_result()def read_result(self):self.euks = self.read_eukfile(self.eukout)self.bacs = set()if self.bacout is not None:self.bacs = self.read_eukfile(self.bacout)def read_eukfile(self, path):lst = set()with open(path) as infile:for line in infile:lst.add(line.strip())return lstclass bin:def __init__(self, path, eukrep):self.e = eukrepself.bname = os.path.basename(path)self.path = os.path.abspath(path)def __str__(self):return "{} euks: {} bacs: {}".format(self.bname, self.table["euks"], self.table["bacs"])def stats(self):"""read bin content and figure genomic composition"""logging.debug("Loading bin")fa_file = Fasta(self.path)stats = {"euks": 0, "bacs": 0, "NA": 0, "sum": 0}# loop and compute statslogging.debug("Make per bin stats")for seq in fa_file:if seq.name in self.e.euks:stats["euks"] += len(seq)elif seq.name in self.e.bacs:stats["bacs"] += len(seq)else:stats["NA"] += len(seq)stats["sum"] = sum([v for k, v in stats.items()])self.table = statsdef decide(self, eukratio=0.2, bacratio=0.1, minbp=100000, minbpeuks=1000000):"""rule to handle decision logic"""keep = Trueallb = self.table["sum"]if self.table["euks"] < minbpeuks:keep = Falselogging.info(f"Eukaryotic DNA amount only {self.table['euks']} instead of target {minbpeuks}")elif self.table["euks"] / allb <= eukratio:keep = Falselogging.info(f"Eukaryotic DNA ratio not higher than {eukratio}")elif self.table["bacs"] / allb >= bacratio:keep = Falselogging.info(f"Bacterial DNA content higher than {bacratio}")elif self.table["sum"] < minbp:keep = Falselogging.info("We did not find at least %d bp of DNA", minbp)self.keep = keepif __name__ == "__main__":parser = argparse.ArgumentParser()parser.add_argument("--output", help="path for the output table", default="assignment.csv", type=str)parser.add_argument("bins", nargs="+", help="all bins to classify", type=str)parser.add_argument("--threads","-t",type=int,help="How many bins should be run in parallel (Default: 1)",default=1,)parser.add_argument("--minl",type=int,help="define minimal length of contig for EukRep \to classify (default: 1500)",default=1500,)parser.add_argument("--eukratio",type=float,help="This ratio of eukaryotic DNA to all DNA has to be found\at least (default: 0, ignore)",default=0,)parser.add_argument("--bacratio",type=float,help="discard bins with bacterial ratio of higher than\(default: 1, ignore)",default=1,)parser.add_argument("--minbp",type=float,help="Only keep bins with at least n bp of dna\(default: 8000000)",default=8000000,)parser.add_argument("--minbpeuks",type=float,help="Only keep bins with at least n bp of Eukaryotic dna\(default: 5000000)",default=5000000,)parser.add_argument("--rerun", action="store_true", help="rerun even if output exists", default=False)parser.add_argument("--quiet", action="store_true", help="supress information", default=False)parser.add_argument("--debug", action="store_true", help="Make it more verbose", default=False)args = parser.parse_args()# define logginglogLevel = logging.INFOif args.quiet:logLevel = logging.WARNINGelif args.debug:logLevel = logging.DEBUGlogging.basicConfig(format="%(asctime)s %(message)s", datefmt="%m/%d/%Y %H:%M:%S: ", level=logLevel)def evaluate_bin(path):if not os.path.exists(path):logging.error("Can not find file {}".format(path))exit(1)logging.info("Launch on {}".format(path))with tempfile.TemporaryDirectory(prefix="filter_EukRep_") as tmp_dir:logging.debug("Using tmp folder: {}".format(tmp_dir))eukfile = os.path.join(tmp_dir, "euks.fna")bacfile = os.path.join(tmp_dir, "bacs.fna")# EukRep can not deal with Gzipped Fasta files, so we unzip it in case it is a Gzip filepath = gunzip(path, tmp_dir)# Launching EukReplogging.debug(f"Starting EukRep on {path}")eukrep_result = EukRep(path, eukfile, bacfile, minl=args.minl)eukrep_result.run()b = bin(path, eukrep_result)b.stats()b.decide(eukratio=args.eukratio, bacratio=args.bacratio, minbp=args.minbp, minbpeuks=args.minbpeuks)return b# multithreading poolpool = Pool(processes=args.threads)results = pool.map(evaluate_bin, args.bins)pool.close()pool.join()with open(args.output, "w") as outfile:outfile.write("path,binname,passed,bp_eukaryote,bp_prokaryote,bp_unassigned,bp_sum\n")for b in results:outfile.write(f"{b.path},{b.bname},{b.keep},{b.table['euks']},{b.table['bacs']},{b.table['NA']},{b.table['sum']}\n")