🌞欢迎来到机器学习的世界
🌈博客主页:卿云阁💌欢迎关注🎉点赞👍收藏⭐️留言📝
🌟本文由卿云阁原创!
📆首发时间:🌹2024年3月15日🌹
✉️希望可以和大家一起完成进阶之路!
🙏作者水平很有限,如果发现错误,请留言轰炸哦!万分感谢!
目录
新药研发
分子对接原理和应用
分子对接的概念
分子对接的原理
分子对接的一般过程
分子对接的分类
分子对接的问题和代表对接软件
打分函数
虚拟筛选流程
尚未解决的问题
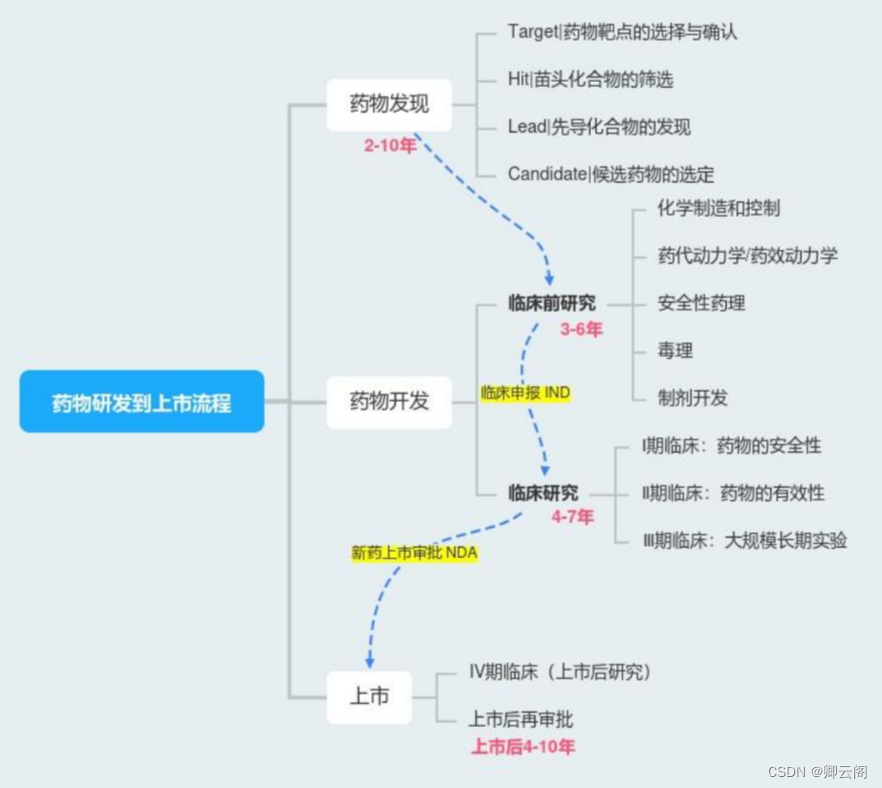
新药研发
药物发现Drug Discovery
药物靶点(Target)的选择与确认,苗头化合物(Hit)的筛选(随机高通量筛选,虚拟筛选),先导化合物(Lead)的发现(从多个苗头化合物中, 决策出活性最好的一个(或几个)作为先导化合物用于继续深入研究)候选药物(Candidate)的选定。
药物开发Drug Discovery
上市(新药上市审批(New Drug Application, NDA)等等)
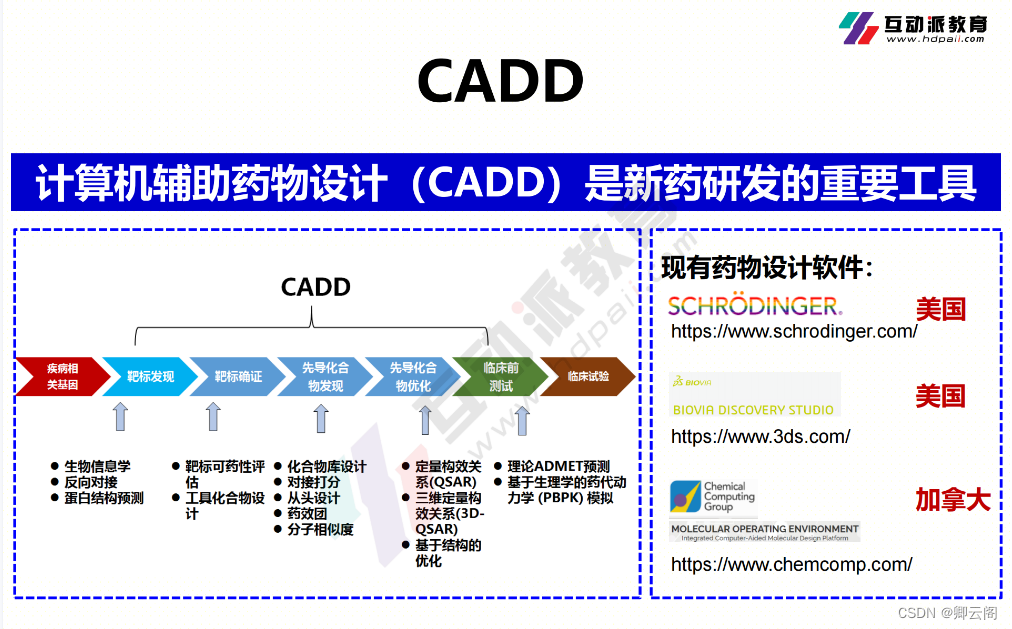
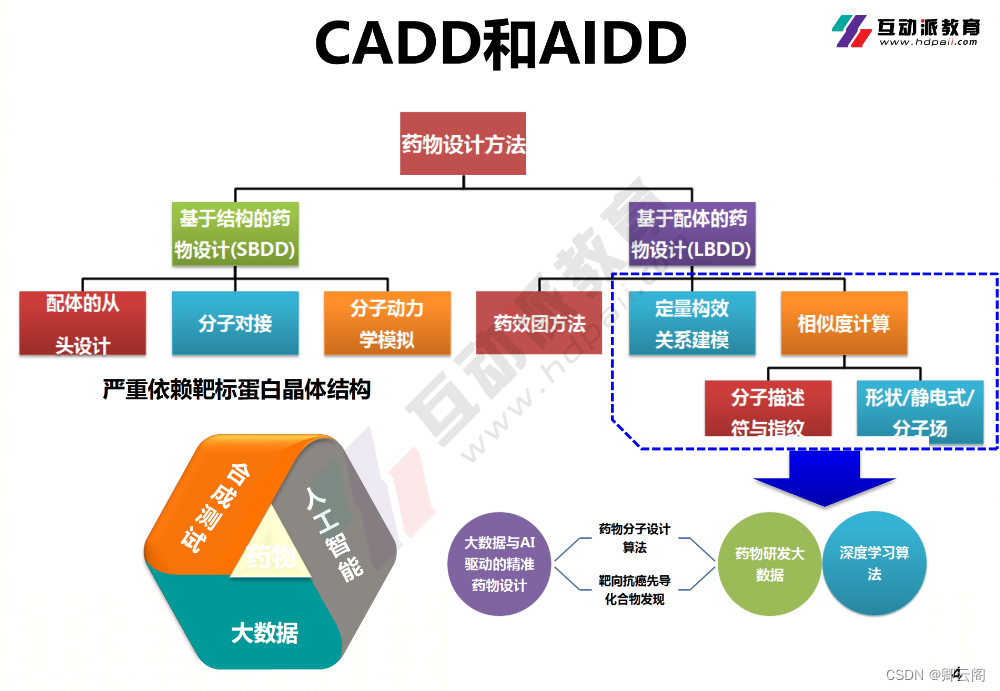
随着科技的发展,我们可以利用新兴手段去提升新药研发到上市的效率,比如计算机(CADD)和人工智能(AIDD)。现有的药物设计软件,基本都是被国外垄断。
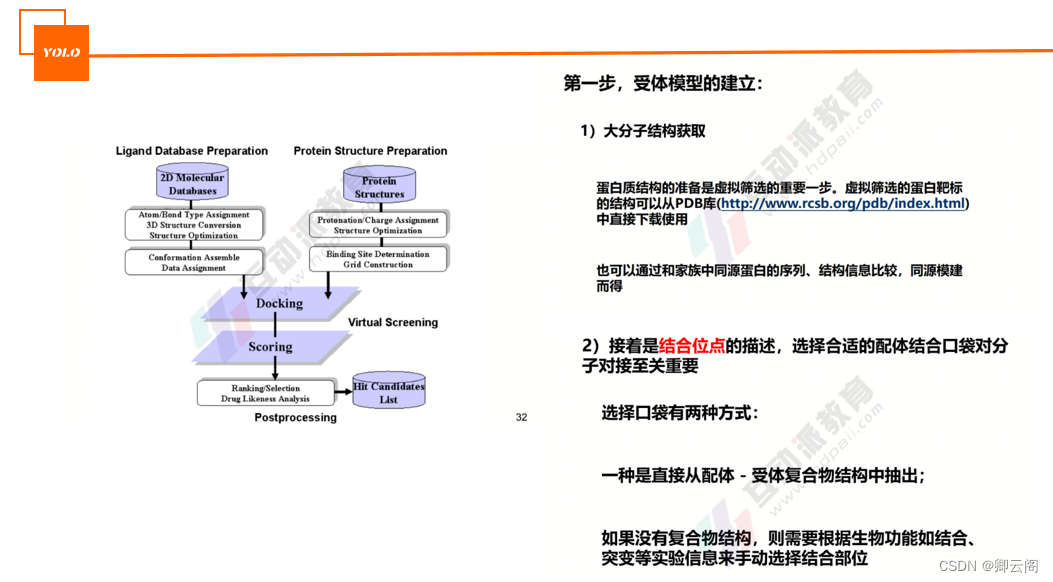
基于结构的药物设计(SBDD):在已知生物大分子靶点结构的情况下,直接考虑药物与靶点的相互作用来进行药物设计。
基于配体的药物设计(LBDD):在生物靶点结构未知的情况下,通过研究与靶点具有特异性结合配体的结构信息,发现先导化合物的方法。
AI主要在定量构效关系建模和相似度计算方向应用的比较多。
分子对接原理和应用
分子对接的概念
分子对接是通过受体的特征以及受体和药物分子之间的相互作用方式来进行药物设计的方法。
受体和药物分子之间通过空间匹配和能量匹配而相互识别形成分子复合物。
空间匹配:假设我的蛋白口袋很小,药物分子是很难进入这个口袋。
能量匹配: 蛋白分子和药物结合精度越低越好,越低说明其处于最低的能量状态,结合比较稳定,比如化学反应的话,一定处于激发态,才会释放能量。代表蛋白质和小分子之间的结合力越牢靠。
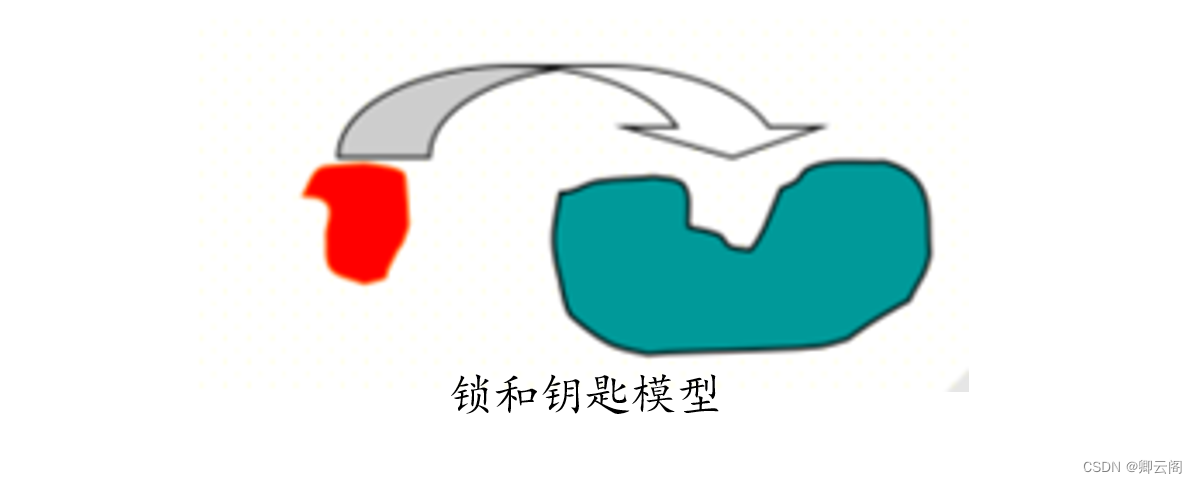
分子对接的原理
锁和钥匙模型
诱导契合模型
药物设计的最终目的:设计一个小分子,可以很好的进入我所设计的靶标口袋里面,并且待的时间越久,效果越好。
分子之间结合自由能,由这几部分组成的,药物和蛋白质之间到底结合的好不好,主要由焓变和熵变两部分组成,熵变的计算是很慢的,但是分子对接不能很慢,所以我们一般忽略熵效应,焓效应也是做忽略计算,最后只是计算范德华,氢键作用等。有些打分函数是把氢键作用放到范德华力里面的(结合自由能,一般都是比较粗糙的,比如忽略人体的水溶液环境),计算效率和计算精度要平衡。
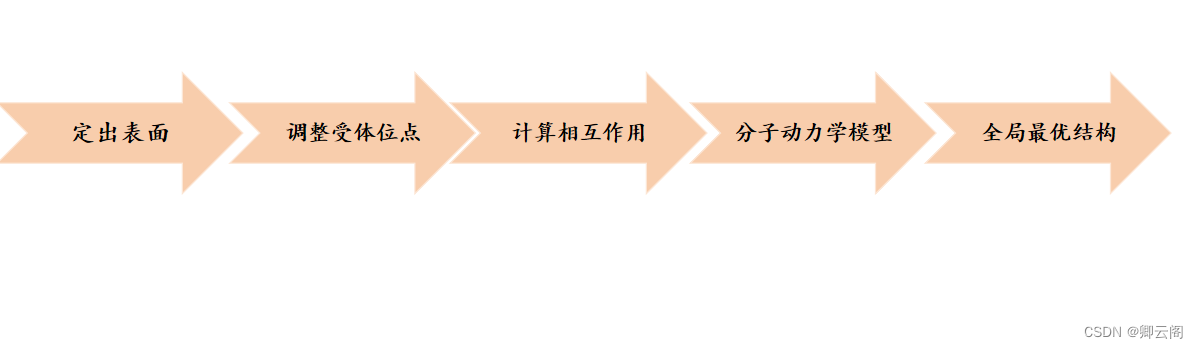
分子对接的一般过程
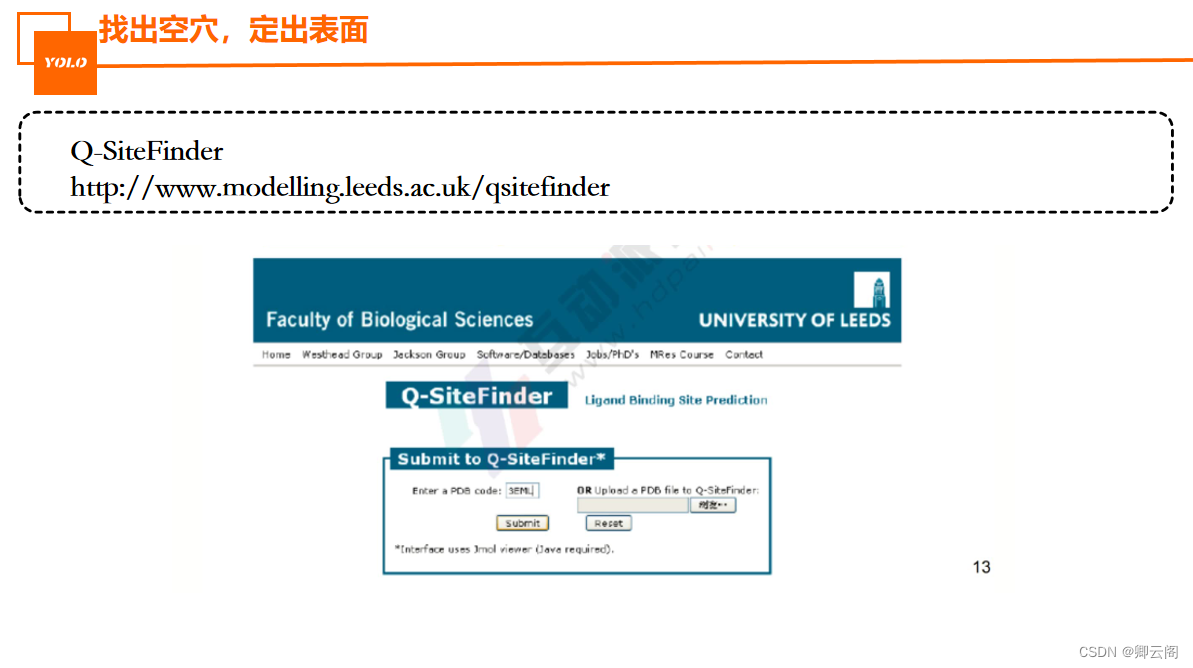
蛋白口袋都是非规则的,(在地面挖个洞怎么把这个体积算出来,我们可以向里面灌水),类似的我们可以用原子进行填充,让其在口袋里滚动,这样就可以根据原子的个数和轨迹计算出体积,一般是全新的才需要找口袋。(一般都是原始晶体复合物占据的位置)
分子对接的分类
刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。
半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。
柔性对接:柔性对接方法在对接过程中允许研究体系的构像发生自由变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,适合精确考察分子间识别情况。
分子对接的问题和代表对接软件
打分函数
虚拟筛选流程
尚未解决的问题
【1】不看后悔系列:只有药物设计能凑齐的绝版阵容! (qq.com)
【bioinformation 6】分子对接










本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.hqwc.cn/news/541715.html
如若内容造成侵权/违法违规/事实不符,请联系编程知识网进行投诉反馈email:809451989@qq.com,一经查实,立即删除!相关文章
APP自动化测试-Appium Inspector入门操作指南
上一篇博客APP自动化测试-入门示例-CSDN博客介绍了APP自动化测试的入门示例,下面详细介绍下Appium 实现的页面元素查看器工具:Appium Inspector的使用方法。
Appium Inspector简介
Appium Inspector 是 Appium 测试框架中的一个工具,用于可视化和调试移动应用程序的 UI 结…
苍穹外卖问题记录(持续更新)
Day01_3.2.4前后端联调
1. 前端无法登录
(1)确保nginx服务器已经启动 (2)查看自己数据库的用户名和密码是否和老师的一样,不一样的话需要在application-dev.yml文件中把老师的用户名密码修改成自己的
老师的用户名…

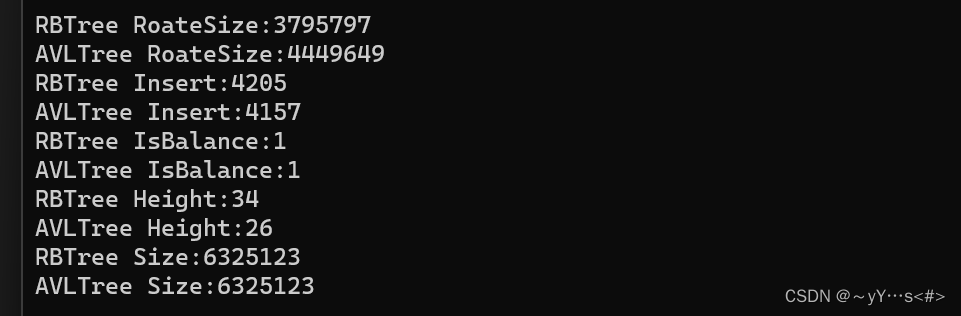
【C++】实现红黑树
目录 一、认识红黑树1.1 概念1.2 定义 二、实现红黑树2.1 插入2.2 与AVL树对比 一、认识红黑树
1.1 概念
红黑树是一个二叉搜索树,与AVL树相比,红黑树不再使用平衡因子来控制树的左右子树高度差,而是用颜色来控制平衡,颜色为红色…
VS2022一个项目中运行多个c++程序
VS2022一个项目中运行多个c程序设置 问题情况解决 问题
一般使用vs2022都需要配置好一些路径依赖,但一个项目中只能使用一个源文件,这也是为了避免找不到那些依赖,可是我们就是想为了可以快速编写,而不是浪费在那些配置环境的时间…

面试常问:你在项目中遇到了哪些比较棘手的问题?怎么解决的?
你在项目中遇到了哪些比较棘手的问题?怎么解决的?这个问题是面试官经常会问的一个问题。
如果你回答我在项目中没有怎么遇到,那么面试官会觉得你什么都不会,对项目了解也不够深入也没有负责什么项目。
面试官其实还挺关心的是应聘者的问题…

【IJCAI】CostFormer即插即用的MVS高效代价体聚合Transformer,FaceChain团队出品
一、论文题目:
CostFormer: Cost Transformer for Cost Aggregation in Multi-view Stereo,https://arxiv.org/abs/2305.10320
二、论文简介: 多视角立体是三维重建的一种重要实现方式,该方式会从一系列同一场景但不同视角的二维…

Navicat破解 Navicat下载安装 附教程 免费
百度网盘:https://pan.baidu.com/s/1wRRN_18_uXxPiIWCS4l43A
麻烦各位师傅帮忙填写一下问卷,提取码在问卷填写结束后显示~ 【https://www.wjx.cn/vm/mBBTTKm.aspx# 】 (资料来源于网络,侵告删)

【Spring IOC/DI】bean 的 5 种注册 与 5 种注入
什么是 bean 一个 bean 就是一个实例化对象 User user new User()
上面这行代码中的 user, 就是 User 类的实例化对象,即一个 bean(User Bean)
什么是 IOC Inversion of Control 控制反转(反转对 bean 的控制&#…

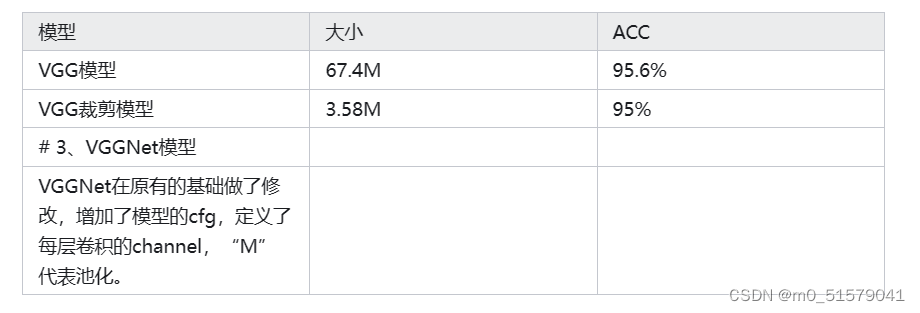
【剪枝实战】使用VGGNet训练、稀疏训练、剪枝、微调等,剪枝出只有3M的模型
摘要
本次剪枝实战是基于下面这篇论文去复现的,主要是实现对BN层的γ/gamma进行剪枝操作,本文用到的代码和数据集都可以在我的资源中免费下载到。 相关论文:Learning Efficient Convolutional Networks through Network Slimming (ICCV 2017…