文章目录
- 摘要
- 正文
- 量子计算现状
- 挑战与前景
- 展望
摘要
量子计算机的工业应用通常取决于其执行准确、高效量子化学计算的预期能力。计算药物发现依赖于对候选药物在有限温度下涉及数千个原子的细胞环境中如何与其靶标相互作用的准确预测。尽管量子计算机还远未被用作制药行业的日常工具,但在这里,我们探讨了将量子计算机应用于药物设计的挑战和机遇。我们讨论了这些可以改变工业研究的地方,并确定实现这一目标所需的实质性进一步发展。
正文
自 1950 年代以来,制药业的药物开发成本从数千万美元增加到数十亿美元,即使根据通货膨胀调整数据也是如此1.为了在治疗未满足的医疗需求方面保持进展,必须寻找药物开发方法的每一个改进来源。作为这一战略的一部分,计算方法在研发中发挥着越来越大的作用2,3.
所采用的方法包括经典分子动力学的模拟技术4,5到量子力学计算6,还有更通用的工具,如机器学习7,8.然而,预测化学系统的行为,特别是量子力学效应,可能是计算密集型的,而且其中许多方法缺乏实际应用所需的速度和准确性。
有人提出,量子计算机可以通过利用量子力学特性来有效地模拟量子系统9,10.这是量子优势的一个例子,量子计算机的预期能力优于经典计算机。受这一承诺的启发,量子计算研究近年来激增,在量子硬件和算法方面都带来了实质性的改进11,12.这些最近的发展吸引了来自私营和公共部门的投资,通常以寻找实际应用为目标。这些投资最常被引用的理由之一是应用量子计算机来增强量子化学计算12、13、14.
目前量子计算在化学中的应用侧重于为最具挑战性的电子结构问题寻找算法,其中电子之间存在很强的相关性。这些影响并不总是被经典计算机可访问的计算完全捕获,因此可以预期与经典计算相比的最大优势。然而,识别具有强电子相关性的系统是困难的15,而且指标数量有限(插文1)16、17、18、19、20、21、22、23、24、25、26、27.尽管解决电子结构问题是许多化学应用的重要步骤,但如果量子计算机的优势仅限于当前技术无法访问的系统,那么它们在计算机辅助药物设计中的实际适用性可能有限。
在这个视角中,我们重点关注未来量子计算机对与制药行业相关的化学计算的适用性。在我们看来,这些将是影响药物设计的第一个可行应用。当然,量子计算机也可能以其他方式影响药物发现,例如,通过增强机器学习协议、分子特性预测或“化合物生成”28.要实现实际适用性,需要大量的发展,需要进行独立、深入的讨论。此外,目前尚不清楚机器学习算法的量子优势是否可以应用于制药相关应用。数据表示方面存在未解决的问题,一些最初有前途的方法已成为“去量子化”的牺牲品,即将量子算法改编为同样有效的经典算法29,30,31,32.
在确定了量子化学中的问题后,量子计算机可以与经典计算方法相比提供加速,我们将这些问题与计算机辅助药物设计中的实际计算需求进行了比较。最后,我们讨论了使量子计算机成为制药行业必不可少工具的研究方向。
插文1 一些具有强电子相关性的指标
量子计算机有望为解决强相关系统的电子结构问题提供优势。在这里,我们报告了五个不同的强电子相关性指标16,17:多重参考:系统的基态需要许多具有可比振幅的参考态(行列式)18,19,20.基本自旋对称性破坏:自旋对称性破坏不是通过添加动态相关性来固定的21.集群扩容失败:当集群扩容方法具有特征性故障点时,说明需要多参考模型22,23.近简并自然轨道:例如,从轨道占据分析中检测到非整数占据数18,24,25.纠缠轨道的数量:这与系统大小成正比,系统大小足够大,传统上很难26,27.
量子计算现状
量子计算领域在过去十年中发展迅速10、11、12.然而,实用的量子优势,即设备产生的工业相关结果超出了经典计算机的能力,仍然需要在硬件和算法设计方面取得重大进展33.
量子算法通常用形成电路的门操作组合来表示(图。1 和 2a)。量子算法发展最重要的指标是计算成本的估算(图1)。2b,c)。这些估计值定义了解决相关问题所需的量子计算资源(量子比特和运行时)。据说涉及许多连续量子门的长运行时间算法具有很高的电路深度。分析解决问题所需的电路尺寸和深度为量子硬件提供了具体的工程目标,并阐明了算法的哪些方面需要改进。
图1:量子计算机上的电子结构计算工作流程。

第一步包括经典预处理,以优化我们想要模拟的哈密顿量。接下来是量子计算所需的量子电路的生成。量子计算机上的计算从准备初始状态开始,然后使用 QPE 计算更昂贵的基态能量。初始准备的态由许多本征态的叠加组成,但需要与基态高度重叠。测量基态 (GND) 能量时,初始态被投影到基态中,如图所示。根据获得的估计值,可以评估系统的化学性质。
全尺寸图像
图2:量子计算、量子算法设计和量子算法缩放所需的元素。

a,运行量子算法,显示为量子计算堆栈。量子计算所需的元素按层次结构顺序显示。在顶部,显示了抽象算法级别。然后,堆栈从量子电路到逻辑量子门,通过量子纠错实现,再到具有物理量子比特和门的硬件97.b, 量子算法设计。显示了设计量子算法时通常需要的一些示例步骤。此过程首先要找到问题的合适描述。该问题被映射到量子计算机中(例如,使用Jordan-Wigner变换来描述使用量子比特的费米子系统),并定义协议的输入/输出(例如初始状态和基态能量)及其步骤,其中可以包括特定量的读出。然后估计和优化性能指标,包括执行时间和准确性。在运行之前,需要通过用基本量子逻辑运算来描述算法来编译量子电路。然后,可以估计所需的量子比特和门数。c, 量子算法的缩放。Top:电路的计算成本是根据所需的资源来定义的,即量子比特数(电路宽度)和运行时间t(电路深度)。底部:经常研究算法的计算成本作为所解决问题大小的函数的缩放。显示了计算资源的多项式和指数缩放与问题大小的示例。面板 A 经许可改编自:左下角图片,Erik Lucero;右下角的图片,参考文献。46, 施普林格自然有限公司
今天,唯一可用的量子计算硬件是所谓的嘈杂中尺度量子(NISQ)制度,以其嘈杂的性质和有限的量子比特数量而得名。噪声的存在会在尝试的计算中引入错误,从而限制了可以实现的规模和规模。大多数 NISQ 算法(例如变分量子特征求解器)严重依赖经典优化启发式方法,并且实际运行时间(完成计算所需的时间)难以估计34,35.此外,最近的结果表明,在NISQ方法中,实现给定误差所需的测量次数与电路深度呈指数成比例36,37.
由于这些原因,我们在这里专门讨论容错量子计算机。这些利用量子纠错技术来产生对噪声具有鲁棒性的逻辑量子比特38,代价是大量额外的量子比特和运行时间。例如,模拟一种经典的具有挑战性的分子,如铁钼络合物FeMoco,它在固氮中起着至关重要的作用39,估计需要大约 2,000 个逻辑量子比特,这将使用 400 万个物理量子比特实现40.这远远超出了当前量子硬件所能实现的范围35.
量子计算机有望解决电子结构问题,并为所有可处理的经典方法都失败的强相关系统找到基态能量。然而,识别这些系统可能非常苛刻和耗时,并且严重依赖化学专业知识。
在过去的 20 年中,已经开发了几种技术来研究各种从头开始方法如何以及何时失败,从而提供强相关性的指标26,如方框1中报告的那些。其中许多问题出现在多金属系统中,其中多个金属离子处于相似的电子环境和相互作用中。这些出现在一些生物系统中,包括酶(例如,FeMoco39和 P45020),但目前尚不清楚它们有多普遍,也不清楚准确系统描述的附加值是什么。
理论上已经证明,如果提供接近基态的初始态,量子计算机可以在多项式时间(相对于系统大小)内解决电子结构问题,而无需进行任何不受控制的近似41,42.该方法使用量子相位估计 (QPE),这是一种非常有效的算法,用于查找哈密顿量的特征态和特征值,这是许多量子计算方法的核心。
图 1 显示了量子计算机上电子结构计算的工作流程。第一阶段使用经典计算机来细化化学系统的几何形状,确定系统的良好初始状态,并合成要在量子器件上运行的纠错电路。量子计算首先在量子计算机内部准备经典确定的初始状态。工作流程的下一步是将QPE应用于初始状态,以获得基态及其能量。
估计正确基态能量的成本直接取决于初始态与基态的重叠。随着与正确基态重叠的减少,它变得越来越昂贵15,41,42.对此工作流的修改允许计算其他可观察对象43,例如分子力44.
抽象量子算法如何在容错设备上实现通常用分层技术堆栈来描述(图 1)。2a). 这个堆栈的底部是硬件——物理量子比特和门。在这个级别上,门是通过直接控制物理量子比特来实现的45,46.在第二层中,错误通过量子纠错码进行检测和纠正,量子纠错码将量子信息编码为多个物理量子比特38.这些允许实现量子逻辑门,这些门可以组合在一起形成电路,这些电路是量子算法子程序的核心,在顶层表示。在图中。2b:我们报告了设计算法并将其编译为量子电路所需的一些主要步骤。
选择算法的一个关键考虑因素是其缩放性(图 1)。2c),特别是与经典计算机的性能相比。如果所需的计算资源(运行时间和物理量子比特数)与问题的大小按多项式成比例,则算法被认为是有效的。如果对于特定问题,量子计算成本是多项式的,但最好的经典等价物是指数的,那么我们就说指数量子优势。当存在这种优势时,人们相信量子计算机可以解决对于经典方法来说不切实际的问题,甚至考虑到构建和操作量子设备的额外开销。
尽管容错量子算法还不能执行,但存在许多方法来评估其计算成本。例如,对估计FeMoco辅因子的基态能量的任务进行了深入研究19,39.对于该系统,通过算法改进,运行时间估计已从几年减少到几天14,39,40,47.随着算法和硬件的进步,将有可能执行此类计算。然而,鉴于目前多种技术竞相开发大规模量子计算机的格局,每种技术都有自己的限制,很难预测第一次容错计算何时可行。在接下来的章节中,我们将讨论药物设计的最新技术,以及量子计算机可用于解决相关药理学系统问题的电子结构部分14,20,48.
计算机辅助药物设计
制药行业生产的化合物是长期发现和改进过程的结果。主要步骤总结在图中。3a. 药物发现过程从确定与疾病病理学有关的靶蛋白开始。该靶点的药理学调节被认为有利于治疗该疾病49并通过分子与靶标结合来实现。确定口服候选药物(首选的药物给药形式)需要很长时间,从结合非常弱的分子开始,需要几年的优化才能获得有效和安全的化合物1,3,8.
图3:靶向蛋白质的药物设计。

a、药物发现过程中的工作流程3,51.一旦选择了生物靶标,该过程就开始识别与其结合的分子。此命中识别发生在 10 的潜在空间内60分子50,产生数以万计的潜在化合物。从这组产品中,选择具有最佳测量和预测性能的代表,在设计、分析、合成以及计算机和体外测试的重复循环中进入优化阶段。只有极少数高度优化和安全的分子(即那些从计算机模拟到体外和体内测试都显示出良好疗效且无毒性作用的分子)进入临床试验的开发阶段,并且只有一种分子最终被选中由医学机构批准51.在药物设计过程的不同阶段采用的计算方法来自参考文献。3列在右边。b,特定配体(PDB 2RGU;利格列汀,一种用于治疗糖尿病的人类药物)与其靶蛋白的药物结合事件的示意图。考虑了配体的构象/几何形状和取向的集合(左)。配体朝向靶标的一些方法会导致结合,而另一些则不会——如箭头(右)所示。最终,对溶液中未结合和结合结构的集合进行采样(每个都需要计算能量)产生药物-靶标结合的自由能52.
全尺寸图像
最初从 10 种化合物中筛选出数百万种化合物60潜在分子50.在工艺的初始阶段,必须优化许多不同的属性,例如结合亲和力。因此,在所谓的先导化合物和先导化合物优化方案中,在确定下一步临床开发的合适候选药物之前,需要合成数千个分子51.
每个合成的分子都经过体外生化、生物物理和细胞测试。然后使用体内(在生物体中)测定法评估具有良好特性的最佳候选物。目标是获得具有尽可能少优化周期数的临床候选药物。在药物发现的这个阶段,计算方法可以提供见解,并帮助指导适当分子的设计。最近,有报道称,计算设计取得了一些惊人的成功2,8.
如今,计算化学在药物设计中大量使用有两个主要领域。第一种是使用电子结构方法计算化合物与靶标的结合强度或结合亲和力,这是候选药物最重要的特性之一6,52.第二个是药代动力学特性的预测,它决定了化合物如何被吸收、分布、代谢和排出体外。后一种计算通常利用机器学习模型,这些模型是在制药公司早期项目中获得的大型实验数据库上训练的7,53.如上所述,量子机器学习的前景目前尚不清楚。尽管增强预测药代动力学特性的方法对于药物设计很重要,但这需要克服一系列非常不同的挑战,并且需要单独进行广泛的讨论。因此,我们在这里重点计算结合亲和力。
结合亲和力相当于药物与靶标之间的结合自由能。它直接对应于靶点所需的药物浓度,从而决定了药物疗效。这转化为预计的人类或动物治疗剂量,这是药物设计过程中最重要的单一参数。同样,药物不得干扰基本的生物过程,也不得与特定的“抗靶标”位点结合。确定对抗靶标的结合亲和力可以深入了解候选药物的潜在副作用。因此,在化合物优化中,结合强度的计算必须准确54.
不幸的是,基于经典力场分子动力学模拟的最新方法并不能产生可靠的结合强度预测55.目标是实现高精度 — 在 1.0 kcal mol 以内−1的实验结果 — 因为在生理温度下,偏差为 1.5 kcal mol−1已经转化为一个剂量估计,错误了一个数量级。
在原子尺度上,一个系统可以在经典计算机上处理,对于所考虑的不同系统大小,具有许多不同级别的近似值(框2)。与经典力场相比,基于量子力学的密度泛函理论(DFT)或耦合簇方法可以更好地描述分子相互作用,但计算成本要高得多6.
这些计算中的其他困难源于化合物性质的热力学性质56.例如,分子可以通过许多不同的方式与蛋白质结合54和热波动意味着系统可以访问不同的几何形状和结合途径。人们需要确定具有最小自由能的构型——统计学上最常观察到的构型。必须计算许多不同的配置,因此必须计算许多单点计算(图 1)。3b)。
在筛选许多具有相似化学结构的化合物时,直接计算化合物之间结合亲和力的差异通常比完全计算每种化合物然后进行比较要快。这项任务通常通过所谓的炼金术扰动方法来完成52,其中已知化合物逐渐演变成新化合物,相应地调整电子结构57.然后根据差异计算相关的热力学性质。这种集成特性计算通常需要实现药物-靶标复合物的自然时间演化58.
药物与其靶标相互作用的完整描述涉及数千个原子(框2)。自由能计算需要数十亿次单点计算,其中执行能量和力评估(图 1)。此外,在模型中必要地包含明确的溶剂(水)可以大大提高自由度和复杂性56,通常使运行时间变得不切实际。使用力场方法在经典计算机上计算小分子与其靶蛋白的结合自由能可能需要数小时。
例如,通过引入DFT方法,包括量子效应的模拟,使计算成本增加了几个数量级,使得自由能计算的完整DFT处理成本高得令人望而却步。具有更高精度的方法,例如耦合集群技术,需要更多的计算资源,因此完全超出了范围,只能应用于小型系统。
为了克服这些局限性,应用多尺度方法将原子尺度连接到细胞尺度59.为了准确描述分子机理,使用了量子力学/分子力学 (QM/MM) 方法,其中仅用更精确的技术表示催化中心或感兴趣区域,并使用具有成本效益的 MM 方法(如力场)模拟周围区域。尽管在定义QM和MM区域之间的边界条件方面存在挑战,但这些方法是研究大型系统最有前途的方法59.
量子电子结构计算在药物开发中的其他潜在用例包括反应条件的优化60,61用于药物合成和计算分子光谱,用于核磁共振、红外 (IR) 或振动圆二色性 (VCD) 光谱,以识别结构62,63,64,65.然而,药物合成成本通常不是非仿制药市场价格的主要驱动因素。相反,它们是由许多失败的优化计划和临床试验推动的51.此外,对于核磁共振波谱的预测,DFT等低精度方法已被证明在许多情况下都能取得良好的结果62,66
总之,药物设计中的量子力学计算将从DFT和耦合簇方法的加速中受益最大。目前,这些方法为大多数相关系统提供了良好的准确性,但对于药物开发中的广泛应用来说太慢了。这是因为大多数口服药物都是小的闭壳有机分子,需要穿过肠壁才能被吸收。这些分子通常缺乏很强的相关性,并且由于其一般的元素组成,可以用较低精度的方法进行处理67.
然而,有一些具有金属中心的药物分子的例子可能在其电子结构中显示出一些强烈的相关性,例如,为癌症治疗或组织的对比增强成像而开发的药物68.一个悬而未决的、未被探索的问题是,这种潜在强相关药物的稀缺性是否是由于含金属药物的一些内在的不需要的特征。这可能是由于它们不受欢迎的药代动力学行为或潜在的毒性引起的,使它们不适合作为药物。另一种可能性是,由于计算优化方面的挑战,它们被避免了,这是量子计算机可能有助于克服的障碍。
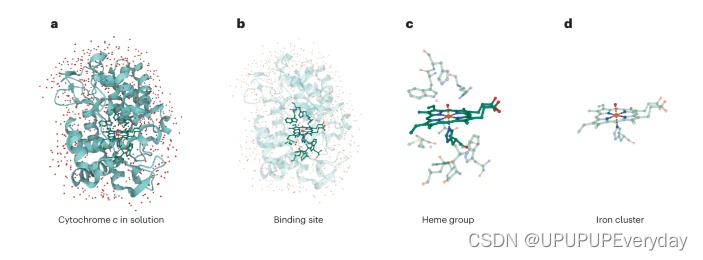
专栏2 常用的化学经典计算方法
从左到右,我们放大细胞色素-c过氧化物酶(PDB 1ZBZ)的化合物I中间体)98.用于对此类系统进行建模的经典计算方法的准确性可能会有很大差异。有些方法可以以较低的精度为代价处理较大的系统,而最准确的方法只能描述非常小的分子。随着该方法精度的提高,经典硬件可以计算出越来越小的分子(这里,从左到右)。a,力场/半经验方法可以模拟大型系统,如溶液中的蛋白质,如细胞色素-c。然而,这些方法不能完全描述量子力学效应,尽管从量子力学方法获得的信息可能会纠正这一点。b,为了模拟蛋白质的中心部分,可以利用Hartree-Fock/DFT方法。平均场法在其他电子的平均电位存在的情况下处理电子。DFT 包括电子相关,而 Hartree-Fock 方法则不包括。 c, 耦合簇 (CC) 方法可以实现更高的精度,即围绕单个平均场参考扩展的簇波函数方法。d, 全配置交互(FCI)方法达到最高精度水平。这种方法在有限的基集中提供了电子结构问题的精确能量,但只能处理少数原子。
挑战与前景
目前量子化学在药物设计中的局限性要么是由于相关系统缺乏准确性,要么是由于包含分子集合的量子效应的可访问方法的计算成本高昂。量子计算机并不能立即解决这两个限制,尽管有希望的想法开始出现。
目前,量子计算机有望通过已知的量子算法(如量子相位估计)加快强相关系统的电子结构计算。例如,这可以用来更好地理解细胞色素P450等酶的物理特性20,这对药物代谢很重要。然而,如果能够超越计算强相关系统的单点能量,那么最大的影响将会出现,因为通常需要热力学特性。
即使经过 30 年的硬件和算法设计的巨大改进12、38、39、45、60、69、70、71、72、73,要使量子计算在药物发现中实用,还需要更多的进展。早期的容错量子计算机很可能无法运行有用的计算,除非算法的设计和实现能够大大提高效率。
量子纠错是执行容错量子算法在空间和时间方面开销成本的主要来源之一。纠错需要每个逻辑量子比特的数千个物理量子比特70,产生数百万个量子比特来计算 P450 的基态能量20.为了减少这些开销,不仅需要开发具有更低错误率和更高量子比特连接性的更好硬件,而且还应该探索对量子纠错的进一步改进38,69.
算法方面的核心挑战之一是在量子计算机上准备一个足够接近目标本征态的初始状态。这是必不可少的,因为找到目标特征态的QPE的运行时间直接取决于初始状态和特征态之间的重叠。随着所研究系统规模的增加,希尔伯特空间呈指数级增长,并且更难准备非常接近目标本征态的初始状态15,74.尽管通过进行算法改进,运行时间随着时间的推移而减少41,42,无法规避对初始状态和目标状态重叠的依赖41.已经提出了几种启发式解决方案9,48,74,但需要进一步研究才能充分了解问题的严重程度。对于不具有强相关性的系统,潜在的解决方案依赖于将系统分解为更小的子系统,并在这些子系统上应用一系列 QPE 以保持整体重叠75.
另一个重要的研究方向是通过解决所需计算的规模或研究特定问题的解决方案来降低总体计算成本,例如找到系统哈密顿量的更紧凑表示,这直接影响量子算法的运行时间40,60,76.同时,与经典算法类似,应该有可能找到基于启发式方法的特定情况的量子算法,这些算法比一般算法的扩展性要好得多。然而,由于缺乏纠错的量子计算机,今天启发式方法的基准测试也得以发展。找到分析这些启发式方法对特定问题的计算成本的方法将有助于理解它们在药物设计中的适用性。
目前的量子算法专注于以最高精度提供加速,这并不总是与工业应用相关。与近似的经典方法相比,运行时间的大幅改进将产生更可观的中期影响。然而,在量子计算机上加速近似技术似乎非常具有挑战性。例如,已经有 DFT 实现随系统大小线性扩展,这使得量子计算机极难超越其性能。
相反,量子计算机可以为系统的物理特性提供新的见解,以提供对经典方法的改进。例如,人们可以使用量子计算来设计更好的DFT泛函。 或者,使用量子计算机来加速收缩张量网络中的经典计算可能是可行的77或者在量子计算机上实现耦合集群技术,这可以在优化阶段实现二次加速78.
最近的研究结果还表明,量子计算机在模拟电子动力学方面可以胜过经典的平均场方法79.将来,人们可以探索其他途径在精度和成本之间进行权衡,例如,通过调整哈密顿模拟的数值精度或截断哈密顿量中的信息量。
尽管单点计算可以深入了解系统的物理特性,但我们通常需要数十亿次单点计算才能确定感兴趣的热力学量,例如结合亲和力。这种大量的计算,每个计算的量子计算运行时间都以天为单位20,44,因此无法在合理的时间内获得结果,更不用说与高度优化的实验的运行时间竞争了。
更实际的热力学量计算的潜在途径可能来自同时在量子计算机上模拟经典原子核和量子力学电子,例如,通过将原子核的经典概率密度函数映射到量子态80.人们可以设想通过生成几何形状的热系综,直接在量子计算机上计算热力学特性,例如自由能81.这是最近趋势的一个例子,量子模拟的优势已经扩展到经典系统80,82,83 元.
另一种选择是使用量子计算机的电子结构计算来优化分子对接的评分函数。此外,细胞依赖性环境因素(如pH值和盐浓度)会影响蛋白质的质子化态和氢键特性,从而对药物设计产生重大影响84,85.未来的量子算法必须隐式或显式地结合这些效应,或者通过嵌入方法在经典计算机上添加它们。以类似的方式,量子计算机可以通过以最高精度处理QM区域来潜在地提高多尺度QM/MM方法的精度,与经典的全配置交互方法相匹配。
除了平衡能量学之外,预测酶速率或药物停留时间等动力学特性还需要使用先进的采样技术,并最终开发更精细的量子算法。在更具推测性的方面,应用于量子计算结果的量子机器学习算法有可能预测药代动力学特性86.当大型量子计算机可用时,我们可能能够计算许多分子集合的波函数,并随后在这些波函数上运行量子机器学习算法87,88.
展望
目前经典的量子化学算法无法准确有效地描述量子系统,无法用于药物设计。只要计算成本低于实验工作量,更准确的计算可以用计算机计算取代许多劳动密集型实验,从而为制药行业带来巨大的价值。可靠的量子模拟可以利用直接从波函数中推导出特性的方法,实现对化学系统的关键、实验上无法获得的见解88.对于这些宝贵的见解,即使在工业环境中,高计算成本也是合理的。
为了对制药行业产生深远的影响,量子计算机需要使更广泛的问题受益,而不是目前经典计算机完全无法解决的少数问题10,12.相关系统有数千个原子(例如大型蛋白质结构及其周围环境),很少需要精确的精度。然而,在许多制药用例中,必须确定热力学特性,这依赖于大型热力学系综,因此需要许多单点计算。寻找新的方法,允许在量子计算机上以准确性换取时间,或者避免采样,这可能是有益的。
理想情况下,量子计算机应该为所有系统提供准确性和鲁棒性,无论是否强相关,其速度目前只有低精度方法才能达到。通过消除经典计算机上的一些当前近似值,药物设计中的全量子计算将变得真正具有预测性,并得到更广泛的应用。
硬件和量子算法的重大进步不断降低计算成本40、60、89、90、91、92、93、94、95.可以理解的是,在不久的将来,特定的量子化学计算可能会从量子计算中受益。然而,现在准确预测制药行业何时将充分利用量子计算的全部潜力用于其应用还为时过早。正如我们所讨论的,这将需要进一步改进硬件和纠错码以及开发新的算法。
已经采取措施解决其中一些挑战,并且有几种想法可以实现这些目标。到目前为止,药物设计的要求与量子算法研究的重点之间存在脱节。这两个学科的科学家都需要了解这两个领域的要求、挑战和局限性。我们相信,只有科学研究以及来自不同领域的科学家之间更强有力的沟通与合作——整合学术界和工业界——将有助于使量子计算成为更快地设计更好药物的重要工具。
Cite this article
Santagati, R., Aspuru-Guzik, A., Babbush, R. et al. Drug design on quantum computers. Nat. Phys. (2024). https://doi.org/10.1038/s41567-024-02411-5