[摘要] 二代测序(next generation sequencing,NGS)已成为中国临床肿瘤医生常用检测工具,而中国超 90%临床医生需要 NGS 报告解读支持。因此,为提升临床医生 NGS 报告解读能力,特编写了 NGS 临床报告解读指引,以帮助临床医生梳理 NGS 报告解读逻辑,快速抓取关键信息,同时尽可能规避过度解读基因组信息导致的潜在危害。本文从临床靶点或驱动基因相关体细胞变异注释及解读、NGS 报告解读及临床决策、可报告范围及质量控制等方面详细介绍了 NGS 报告解读应遵循的恰当的结构化循证原则,正确理解 NGS 报告的逻辑结构、抓取关键信息并综合分析,为肿瘤患者带来切实的临床获益。帮助医生在综合浏览完一份样本检出的所有分子变异后,结合患者的基本临床信息以及既往或同期其他配对样本检测结果,综合判断患者疾病的全面分子特征谱及其演化过程,了解这些信息所提示的生物学意义和临床意义,最终做出正确的临床决策。
[关键词] 二代测序;报告解读;指引
目 前 二 代 测 序(next generation sequencing,NGS)已成为中国临床肿瘤医生常用检测工具,中 国 临 床 肿 瘤 学 会(Chinese Society of Clinical Oncology,CSCO)肿瘤生物标志物专家委员会发布的第 1 个 NGS 临床应用调研显示,大于 30%的肿瘤科医生每月 NGS 检测量超 5 个,而中国超过 90%临床医生需要 NGS 报告解读支持。为了提升临床医生 NGS 报告解读能力,我们特意邀请全国多个临床肿瘤学专家及 NGS 检测专业人员,共同编写了NGS 临床报告解读指引,以帮助临床医生梳理 NGS报告解读逻辑,快速抓取关键信息,同时尽可能规避过度解读基因组信息导致的潜在危害。对于医生来说,能否正确理解 NGS 报告的逻辑结构、抓取关键信息并综合分析,以实际指导临床决策,决定了NGS检测能否为肿瘤患者带来真实的临床获益。
每个肿瘤基因组中可能存在数百至数千个体细胞突变,其中许多是相对个体化的变异。确定通过NGS鉴定出的多个基因变异(genomic alterations,GA)的优先级排序是一项重大挑战。这些基因变异主要包括单核苷酸变异(single ⁃ nucleotide variant,SNV/single⁃nucleotide polymorphism,SNP)、短片段插入/缺失突变(Indel)、重排(融合)、拷贝数变异(copy number variation,CNV)及其他复杂突变等。部分变异出现在生物学及临床相关、甚至是分子治疗潜在靶标的肿瘤基因中,但并非所有肿瘤相关基因发生的变异均为(潜在)功能性变异,更多的基因变异尚无明确的生物学和/或临床意义。随着高通量测序分析进入临床领域,产生了大量数据,而如何及时、准确的将测序发现的肿瘤基因组变异信息转化为临床医生可读取并用于指导临床决策的结构化循证报告(structured evidence⁃based reports),正变得越来越重要。现从以下3个方面对NGS临床报告解读作以指引。
1 临床靶点或驱动基因相关体细胞变异注释及解读
基于 NGS 技术检测肿瘤体细胞变异的实验流程可概括为以下几个主要环节:样本采集及质量控制、DNA 提取、文库制备、测序、基因组数据生成及数据分析。其中,数据分析可进一步拆解为三个流程:变异识别(variant identification)、变异注释及过滤(variant annotation and prioritization)、变异的临床解读(interpretation of clinical significance)。其中,变异识别、注释及过滤经由生物信息学工具实现;而临床解读则需要基于严格的分级逻辑,整合当前公共数据库及已发表文献的海量信息,特别是变异⁃药物敏感性信息,建立基因变异的临床解读知识库,最终将与送检样本的对应癌种及检出的基因变异相匹配的临床意义(如药物敏感性信息)及其证据级别呈现在 NGS 报告中。
目前有多个循证分级系统可用于指导基因体细胞变异的临床解读。包括 2017 年美国分子病理学协会(Association for Molecular Pathology,AMP)/ 美 国 临 床 肿 瘤 学 会(American Society of Clinical Oncology,ASCO)/美 国 病 理 学 家 协 会(College of American Pathologists,CAP)联合制定的体细胞变 异解读指南,2018 年欧洲肿瘤内科学会(European Society for Medical Oncology,ESMO)发布的分子靶点临床可操作性量表(the ESMO Scale for Clinical Actionability of molecular Targets,ESCAT)以及纪念斯 隆 ⁃ 凯 特 琳 癌 症 中 心(Memorial Sloan Kettering Cancer Center,MSKCC)的 精 准 医 疗 肿 瘤 数 据 库 (Precision Oncology Knowledge Base,OncoKB)证 据 等级规则。总体而言,无论哪个分级系统都遵循一些共性原则,包括循证、跨癌种处理等,故其中并无优先推荐者。医生在阅读一份 NGS 报告时应先了解其变异解读依据的证据分级原则及其采用知识库的局限性,以帮助自己更好的理解报告内容。
根据 2017 年 AMP/ASCO/CAP 联合制定的体细胞变异解读指南,体细胞变异在不同癌种中对应的药物敏感性证据分为四个等级:A级,美国食品药品监督管理局(Food and Drug Administration,FDA)批准或专业临床指南推荐;B级,经具有足够统计学效能的临床研究证实、获得该领域专家共识;C 级, 其他癌种中的 A 级证据(跨适应证用药)、或已作为临床试验的入组标准;D级,临床病例报道或临床前 证据支持。体细胞变异对特定肿瘤的诊断及预后价值,亦给出相应分级:A 级,专业指南中定义的特定肿瘤的诊断/预后因子;B 级,经具有足够统计学 效能的临床研究证实其诊断/预后价值;C 级,多项小型研究支持其诊断/预后价值;D 级,小型研究或个案报道提示其辅助诊断/预后价值(独立或联合其他标志物)。
基因变异按照其临床意义的重要性分为四个等级:Ⅰ类变异,具有 A 或 B 级证据;Ⅱ类变异,具有 C 或 D 级证据;Ⅲ类变异,临床意义不明;Ⅳ类变异,已知无临床意义。详见图 1。该体细胞变异解读指南在国内影响范围最广, 许多第三方NGS检测公司的临检报告即遵循该指南的分级原则对基因变异进行解读,详见表1。
ESCAT 是由 ESMO 转化研究和精密医学工作 组(Translational Research and Precision Medicine Working Group,TR and PM WG)发起建立的、基于 临床靶标相关证据的基因变异临床分类系统于 2018 年发表。将基因变异分为六个级别:Ⅰ级, 可用于常规临床决策的靶点,如乳腺癌的 HER2 扩 增 和非小细胞肺癌(non ⁃small cell lung cancer,NSCLC)的 EGFR 敏感突变。其中Ⅰ⁃A 级突变是基 于前瞻性临床研究已有了显著生存获益的靶标突变类型;Ⅱ级,证据表明患者将受益于针对性靶向治疗,但仍需更多数据证实。如 PI3K 通路中的AKT1、PTEN 突变;Ⅲ级,在其他肿瘤类型(而非该患者肿瘤类型)中已证实临床获益的靶标性基因变异。如 NSCLC 以外肿瘤中的 EGFR 19del 突变;Ⅳ级,仅有临床前证据支持靶标性的基因变异;Ⅴ级,有证据表明针对此类变异的靶向治疗可获得客观缓解,但缺乏有临床意义的生存[无进展生存期(progression ⁃ free survival,PFS)或 总 生 存 期(overall survival,OS)]获益,或可支持联合治疗策略;Ⅹ级:已证实缺乏临床价值而非“尚无证据支持”),不应影响临床决策。

OncoKB 是由 MSK 癌症中心维护的精准医疗肿瘤数据库。该数据库旨在提供详细的、基于证据的基因组变异相关信息,以帮助临床医师进行治疗决策。数据基于 FDA、NCCN、ASCO 指南等权威资料及其他科学文献、以及疾病专家小组的建议,对不同变异的药物预测价值进行证据分类:1/2 级,FDA 认可、或被认为是临床标准(standard of care,SOC)生物标志物,可预测在特定疾病背景下对已获批药物反应的变异;3 级,基于对临床试验中待测目标药物的有希望的临床数据,被认为可预测药物反应的变异;4 级,根据对临床试验中待测目标药物的令人信服的生物学证据,被认为可预测药物反应的变异。
2 NGS 报告解读及临床决策
一份结构化循证报告的背后,需要基于特定逻辑建立临床解读知识库框架,并通过对开源知识库及海量科学文献的信息甄别、分级、编辑,不断完善机构内部临床解读知识库。通过生物信息分析流程将基因变异识别、注释、过滤后,可报告变异进入临床解读知识库进行变异、癌种、证据匹配,最终形成可读的结构化 NGS 临床报告。最终对其进行解读及实施临床决策,NGS 报告解读决策树详见图2。

2.1 针对“全阴报告”的解读逻辑
当医生面对一份体细胞变异“全阴报告”(即 一份样本未能检出任何肿瘤体细胞突变)时,应首先考虑以下两点: (1)送检样本是肿瘤组织、外周血循环游离 DNA(circulating free DNA,cfDNA)或其他;结合样本质控信息及患者治疗史,综合判断样本中是否含有足够肿瘤成分,DNA 总提取量和/或肿瘤占比是否可能低于 NGS 的 LOD;(2)选择的 NGS panel(尤其是仅包含数个至数十个基因热点区域的小 panel)是否与患者的肿瘤类型相匹配,即该肿瘤常见变异是否能被该 panel 覆盖。 以上评估可以帮助我们判断,该“全阴”结果是提示“X 基因野生型”可能性大,还是“X 基因状态未知”可能性大。比如一份血检全阴报告,采用 panel 针对泛癌设计、覆盖上百基因的热点区域,对目标癌种常见变异的覆盖度>90%,此时的全阴,很可能仅提示肿瘤全身负荷较低或其他原因(如患者正接受有效的抗肿瘤治疗)导致释放入血的循环肿瘤 DNA(circulating tumor DNA,ctDNA)含量极低、未达检测平台的 LOD。这种“全阴”不能反映肿瘤基因组变异状态,仅能提示“基因突变状态未知”。当然,这种血检全阴报告可以提供突变谱以 外的其他信息,在特定场景下提示一定的预测、预后价值。
2.2 针对 NGS 报告中突变丰度及拷贝数的解读逻辑
医生在拿到一份 NGS 阳性报告时,可能对其中 检 出 的 突 变/重 排 的 丰 度(allele frequency/ fraction,AF)及 CNV 的拷贝数(copy number,CN)存在疑问。基于不同的样本类型,对 AF 及 CN 的理解可能存在一定差异。
2.2.1 基于组织样本的 NGS 检测
首先对 AF 绝对数值的解读。对于组织检测结果,可以基于如下公式,根据 X 基因突变(或重排)的 AF 以及样本中的肿瘤占比,粗略判断该变异在肿瘤中的克隆占比:
X 基因变异 AF = 肿瘤占比 × 肿瘤中 X 变异克 隆占比 ×0.5(杂合突变)[或 1.0(纯合突变)/中性拷贝数 LOH]。
一般情况下,肿瘤细胞中的突变,尤其是癌基因突变(如 EGFR、KRAS 活化突变,ALK、ROS1 重 排)以杂合突变存在;但上述公式未考虑潜在的拷贝数变异(比如 EGFR 19del 伴扩增)的影响。而抑癌基因的功能缺失性(loss of function,LOF)杂合突变,则可能由于杂合性缺失(loss of heterozygosity, LOH)的存在而影响克隆占比的估算。拷贝数为 2 的 LOH(中性拷贝数 LOH)可类比纯合突变进行肿瘤克隆占比的估算,而拷贝数缺失的 LOH 则更加 复 杂 。 例 如 ,一 份 晚 期 肠 癌 组 织 样 本 中 ,检 出 NTRK1重排(不伴 CNV),AF 为 10%;而病理评估提示该样本肿瘤占比约为 50%;根据公式计算(考虑为杂合突变),携带 NTRK1 重排的肿瘤细胞仅占所有肿瘤细胞约 40%,提示 NTRK1 重排仅存在于肿瘤的一个亚克隆。当然,用于病理评估的样本仅为一张切片,不能完全代表检测样本整体;且病理评估还可能受局部肿瘤坏死等因素干扰,故病理评估结果仅供参考,由此推算出来的变异克隆占比与实际情况可能存在一定偏差。而当样本无法进行病理评估时(比如病理蜡卷标本),则只能以该样本中检出变异的最高突变丰度(maxAF)来反推可能的肿瘤占比。比如一份肠癌样本中同时检 出 KRAS G12D 突变、SMAD4 突变和 NTRK1 重排,KRAS 突变作为 maxAF 丰度为 25%,反推该样本肿 瘤占比约为 50%,以此为基础评估 NTRK1 重排的克隆性。
其 次 对 AF 相 对 数 值 的 解 读 。 组 织 样 本 中 不同变异的相对丰度同样可以提供很多信息。 比 如 一 份 肺 癌 组 织 样 本 中 ,同 时 检 出 EGFR 19del 和 ALK 重排,二者相对丰度为 5∶1;结合患者 既 往 EGFR ⁃ 酪 氨 酸 激 酶 抑 制 剂(tyrosine kinase inhibitor,TKI)长期治疗史,我们有较大把握推断,该 ALK 重排为获得性耐药机制,而非 EGFR/ALK 双 原发肿瘤。而大样本研究显示,晚期肠癌患者 cfDNA 样本中检出的可靶向 TRK 重排(融合)多以亚克隆形式存在,即对比主干突变 maxAF 的相对 丰度低于 50%。
最后对 CNV 的解读。CNV 及基因组上部分区域的扩增或缺失。面对NGS一份报告,首先需要注意 CNV 有不同的报告形式,最常见的包括拷贝数(copy number,CN)及扩增倍数(ratio)。前者即一份样本提取的 DNA 混合物(包含肿瘤 DNA 及非肿瘤来源 DNA)中检出的某基因的平均拷贝数(正常 CN 为 2);后者即前者与 2的比值。基于 NGS检测得到 的 CNV 的非校正 CN 或 ratio 值与荧光原位杂交 (fluorescence in situ hybridization,FISH)检测得到的CN值或ratio值之间存在较大差异,详见表2。

在已知肿瘤细胞占比的情况下,可通过以下公式粗略换算目标基因的肿瘤细胞 CNV(即校正 CNV):
报告 CNV = 校正 CNV × 肿瘤占比 + 2 ×(1⁃肿瘤占比)。
假设肿瘤细胞中目标基因CN为20,当肿瘤占比仅为 10%时,实际检出 CN 值仅约为 3.8。由于正常 DNA的稀释作用,当一份样本中肿瘤占比低于 20% (即maxAF<10%)时,CNV的检出敏感性显著降低。
2.2.2 基于外周血 cfDNA 的 NGS 检测
NGS 血检,检测对象是血浆中的 cfDNA。虽然科研领域人们可以通过一些方法(比如根据 cfDNA片段长度的差异)评估 ctDNA 的实际含量,但在实际临床应用及大多数科研发表中,仍以血浆样本中检出体细胞变异的maxAF作为ctDNA含量的替代指标。可针对某一肿瘤患者进行不同临床节点的血检动态监控,以 maxAF反映外周血 ctDNA 含量的动态变化,以此反映全身肿瘤负荷变化、预测治疗效果及生存预后。比如,对于初诊晚期肿瘤患者,基线的ctDNA 含量高低与全身肿瘤负荷相关;而治疗开始后(3~8周)ctDNA 的早期变化趋势可预测当前治疗的总体效果,ctDNA 快速清零预示更长的 PFS 及OS。这种变化趋势比影像学评估更加快速敏感,比如影像学评估同为SD的患者,ctDNA早期清零与长PFS显著相关;免疫治疗假性进展的患者,治疗初期即表现为ctDNA快速下降甚至清零。由于多数肿瘤患者外周血maxAF<10%,所以外周血检测对CNV的检测敏感性明显低于突变、重排等变异形式。
2.3 针对单个变异对比多基因多变异的解读逻辑
根据不同 NGS panel 的可报告范围,一份 NGS阳性报告中的变异可能有多有少。对于不同情况,临床医生应采取不同的解读逻辑流程。对于多基因变异、特别是多个潜在驱动变异共存的情况,要结合肿瘤类型、既往治疗史、相对突变丰度、既往分子检测结果等信息综合判读,以推测不同变异之间的逻辑关系(原发性 vs. 获得性,主克隆 vs. 亚克隆,敏感克隆vs. 耐药克隆),指导后续治疗。
2.4 胚系突变分级注释
当无配对样本(白细胞或正常组织)时,肿瘤样本(tumor only)的检测在变异注释及过滤过程中,需要建立生物信息学算法,以有效区分肿瘤体细胞突变和胚系突变,确认并过滤胚系多态性。如同时检测配对样本,则可明确区分体细胞突变和胚系突变。对检出的胚系突变,应参照 ACMG 推荐的胚系突变解读流程进行注释及解读。基于当前证据,胚系变异的致病性分为 5级:5级,致病;4级,可能致病;3级,意义未明(VUS);2级,可能不致病;1级,不致病。仅有4/5级胚系变异具有相应的临床指导价值。比如谈及 PARP抑制剂与 BRCA1/2胚系突变的相关性,或BRCA相关卵巢癌⁃乳腺癌的风险评估,均默认仅针对4/5类BRCA1/2突变。
2.5 NGS 可报告的基因组标签
2.5.1 肿瘤突变负荷
肿瘤突变负荷(tumor mutation burden,TMB)即肿瘤基因组编码区包含的非同义突变的数量或密度(突变数/Mb),是肿瘤新抗原负荷的替代指标,过去以全外显子组测序(whole ⁃exome sequencing,WES)作为评估的金标准。目前基于 NGS 大 pane检测的 TMB 已可达到与 WES 的高度相关并在大量免 疫 治 疗 临 床 研 究 中 证 实 其 对 疗 效 的 预 测 价值。目前,业界对组织 TMB 检测有几点共识:①编码区覆盖大于 1 Mb(大概相当于 300 个以上基因的全外显子区域);②基于经过验证的可靠生物信息分析算法。同一份样本采用不同检测平台、不同大 panel、不同的生物信息学算法,都可能导致 TMB 绝对数值的差异。因此,在缺少桥连试验 的 前 提 下 ,直 接 采 用 某 一 家 公 司/产 品(如FoundationOne®CDx)的 肿 瘤 突 变 负 荷 高(TMB high,TMB⁃H)界值(cut⁃off)来定义其他检测产品的TMB⁃H 是不合适的。同时还应注意,对 TMB 的准确评估(无论基于组织或血浆 cfDNA 样本)建立在样本满足一定肿瘤占比的基础上,肿瘤占比过低将导致 TMB 的严重低估。因此,医生在拿到一份NGS 报告,看到 TMB 数值时,应综合考虑以下因素:选择的 panel(有无准确测算 TMB 的能力)、检出的肿瘤突变谱及其突变丰度(肿瘤占比是否足以评估 TMB、突变谱特征与 TMB 高/低的组合是否合理)、TMB 绝对值以及该数值在已检测的肿瘤样本中的相对排序等,综合评估 TMB 水平及其可信度。
2.5.2 微卫星不稳定性
微卫星不稳定性(microsatellite instability,MSI)MSI/错配修复(mismatch repair,MMR)检测对于多种实体瘤患者具有重要临床意义。过去以多重荧光聚合酶链式反应(polymerase chain reaction,PCR)毛细管电泳法作为 MSI 检测的金标准,基于美 国 国 家 癌 症 研 究 所(National Cancer Institute,NCI)推荐的 5~7 个经典微卫星位点,对比肿瘤细胞与正常细胞的检测结果,以确定肿瘤细胞的 MSI状态。近年来,NGS panel 开始用于 MSI 检测,使用计算工具同时研究基因组上的大量微卫星序列成为可能。且多数 NGS⁃MSI 算法采用正常人长度分布模型,无需正常组织作对照。一个 NGS panel需要整合一定数量的有效微卫星位点、构建相应的 MSI 算法并经过临床样本验证,才能准确报告MSI 状态。对 MSI 的准确评估同样也建立在一定肿瘤占比的基础上。对于 MSI检测结果,应综合样本肿瘤占比(可参考肿瘤突变丰度)、TMB 水平[微卫星高度不稳定性(MSI⁃high,MSI⁃H)的肿瘤往往 TMB⁃H]、肿瘤突变谱特征(MSI⁃H 可由 MMR 基因失活性突变导致,且基因突变谱往往以 Indel 为主)等信息综合分析,判断其可信度。
3 可报告范围及质量控制
当临床医生拿到一份 NGS 报告时,应关注其样本主要质控参数、可报告范围(检测内容)、检测方法及其局限性等相关内容,具体示例详见图3 及 表 3。 相 关 内 容 可 能 部 分 位 于 报 告 首 页 后(如检测内容概述),而更多详细信息则以附录形式出现在报告主体内容后,故往往容易被忽略。但一份报告的结果是否真实可信,或者是否能够回答临床医生最关心的问题,恰恰严重依赖于这些看似不起眼的“附加信息”,包括检测方法、可报 告 范 围(检 测 内 容)、质控参数 、局限性说明等。

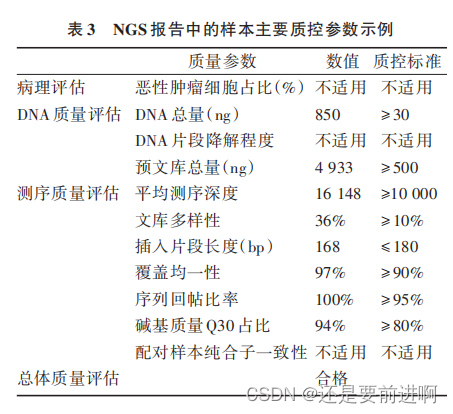
还有一点需要注意,不同变异形式的检出对样本肿瘤 DNA 含量的要求是不同的,换句话说,在相同的 DNA 投入量和肿瘤占比的前提下,NGS 检测对突变、重排、CNV、TMB 及 MSI 状态的检测敏感性不同。比如一份福尔马林固定石蜡包埋 (formalin⁃fixed paraffin⁃embedding,FFPE)样本的肿 瘤占比评估约为 15%(或 maxAF<10%),对于基因突变、重排检测及 TMB 估算来说为合格样本,但 对 CNV、MSI 的检测敏感性则可能受限。此类信 息 的 获 得 依 赖 于 对 报 告“ 局 限 性 说 明 ”的 仔 细 阅读。
NGS 报告端能否遵循恰当的结构化循证原则, 形成可读性较强的临床报告;临床医生能否正确理解 NGS 报告的逻辑结构、抓取关键信息并综合分析以指导临床决策,决定了NGS检测能否为肿瘤患者带来切实的临床获益。无论报告模板如何,无论遵循哪个具体的变异临床解读逻辑,任何一份NGS 报告的解读结果都不应抵触药物说明书或专业临床指南。医生在拿到一份 NGS 报告后,不应拘泥于一条条检测报告结果本身,而应抽提出具体信息,整合到患者的实际临床背景中去讨论。医学强调循证,也强调个体化,临床医生应在综合浏览完一份样本检出的所有分子变异后,结合患者的基本临床信息以及既往或同期其他配对样本检测结果(如果有),判断患者疾病的全面分子特征谱及其演化过程,联系这些信息提示的生物学意义和临床意义后给出临床决策建议,而不是机械地仅仅基于报告检出的证据级别最高的基因变异给出对应用药推荐。对于复杂情况应鼓励采取多学科讨论(如分子肿瘤专家组,Molecular TumorBoard,MTB)的形式辨析不同变异组合的生物学意义和临床意义。
附录:名词解释
· AF/MAF/VAF(allele fraction/ mutation allele fraction/variant allele fraction):突变丰度,或突变/变异等位基因丰度,是指某个基因位点所有的等位基因中,突变的等位基因所占的相对比例,即等于突变型/(突变型+野生型)。
· CEP(chromosome enumeration probe):染色体计数探针,能够与位于着丝粒区域高度重复的alpha 卫星序列结合,用于检测染色体的数目异常。
· cfDNA(circulating free DNA):循 环 游 离DNA,是细胞凋亡或者坏死后降解和释放到外周血的游离 DNA。
· CHIP(clonal hematopoiesis of indeterminate potential):意义未明克隆性造血,是一种普遍的与老化相关的现象,造血干细胞或其他造血祖细胞形成携带一定基因特征的血细胞亚群。这些亚克隆所携带的变异一般丰度较低,可以通过细 胞 等 深 度 测 序 对 照 及 生 信 算 法(部 分)进 行过滤。
· CNV(copy number variation):拷贝数变异,是基因组上部分区域的扩增或缺失。CNV 是一种结构变异,在肿瘤基因组中常见,包括插入、缺失和重复。NGS 可通过对该区域的覆盖度(测序深度)来估算 CNV。
· ctDNA(circulating tumor DNA):循 环 肿 瘤DNA,是外周血中携带一定肿瘤特征(如 SNV、插入/缺失突变、重排、CNV 等),来自肿瘤基因组的DNA 片段。主要来源有凋亡或坏死的肿瘤细胞、循环肿瘤细胞(CTC)以及肿瘤细胞分泌的外泌体。
· LBx(liquid biopsy):液体活检,是指对非固体生物样本(主要是血液)的采样和分析。液体活检和组织活检一样,主要用于癌症等疾病的诊断和监测,与组织活检相比具有无创,能动态检测等优点。
· LOD(limit of detection):最低检测限,是指能以适当的置信度(一般是 95%)被检出的突变的最低浓度。
· LOH(loss of heterozygosity):杂合性缺失,是一种使某特定基因丢失的基因组变化。如果一个特定位点的等位基因一侧正常、一侧有突变,某种原因导致的正常等位基因缺失或突变会产生没有正常功能的基因座。
· MaxAF/MSAF (maximum allele fraction /maximum somatic allele frequency):最大变异丰度,或最大体细胞基因变异频率,是指在组织、血浆等 类 型 样 本 中 ,所 检 出 的 最 高 的 体 细 胞 变 异丰度·。
MMR(mismatch repair):错配修复。DNA 错配修复是指在含有错配碱基的 DNA 分子中,使核苷酸序列恢复正常的一种修复方式。
· MSI(microsatellite instability):微卫星不稳定性。微卫星是指细胞基因组中以少数几个核苷酸(多为 1~6 个)为单位串联重复的 DNA 序列。DNA错配修复(MMR)功能出现异常时,微卫星出现的复制错误得不到纠正并不断累积,使得微卫星序列长度或碱基组成发生改变,称为微卫星不稳定性。
SNV(single ⁃nucleotide variant):单核苷酸变异,是基因组上部分区域发生的点突变。SNV 是最常见的变异类型。
· TBx(tissue/tumor biopsy):组织活检或肿瘤组 织 活 检 ,是 采 集 组 织 并 对 组 织 进 行 检 查 和分析。
· TMB(tumor mutation burden):肿瘤突变负荷,是指肿瘤基因组编码区每百万碱基中的体细胞非同义突变个数。理论上 TMB 越高,可能被 T淋巴细胞识别的新抗原越多,免疫检查点抑制剂疗效可能越好。