1.FastQC的下载,安装
https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc
在这里下ZIP包,这个是已经编译过的,打开设好路径就能用。
注意:FastQC使用之前记得安装好Java Development Kit (JDK),没有这个没有办法工作,会报错。
https://blog.csdn.net/m0_69574256/article/details/133956779
主要参考这篇帖子
主要步骤:
①.wget下载FastQC:wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.12.1.zip
②.解压:unzip 你下下来的包的全名
③.设置路径:echo 'export PATH=/root/FastQC:$PATH'>>~/.bashrc
然后更新一下:source ~/.bashrc
到这里,用fastqc -h测试一下,看看安好了没有,安好了是这样的:

2.FastQC的使用
https://blog.csdn.net/qq_74093550/article/details/131530092
主要步骤:
①.首先,将自己的数据准备好,从哪弄数据看你自己,我这里的数据来源参考先前的博客。

②.fastqc -4 /root/SRA/sratoolkit.3.1.1-centos_linux64/bin/SRR13810477/*.fastq.gz -o /root/SRA/sratoolkit.3.1.1-centos_linux64/bin/SRR13810477/QC
这里用这个命令,其中主要参数如下:
-4:这里指4线程,看你电脑有多少和你打算用多少,自己设
/root/SRA/sratoolkit.3.1.1-centos_linux64/bin/SRR13810477/ :这是你的数据文件的位置,我这么长一串主要是因为我喜欢用绝对位置,这样不容易报错
*:这个指的是你要处理这个文件夹中所有的你要分析的文件
.fastq.gz:这个是你的文件的格式,他支持直接用gz,我也推荐直接用gz,比较省空间,当然也可以直接用fasta、fastq等格式,看自己需求
-o /root/SRA/sratoolkit.3.1.1-centos_linux64/bin/SRR13810477/QC:-o指的是你想把你的结果输出到那里去,我选择新建一个文件夹,好找。
然后你就可以运行了,运行的时候大概是这个样子:
最后获得的结果大概就是这样:
3.结果解读
接下来就是打开它输出给你的网页看图说话了,主要参考这个帖子:
https://blog.csdn.net/qq_44520665/article/details/113779792
https://blog.csdn.net/qq_74093550/article/details/131530092




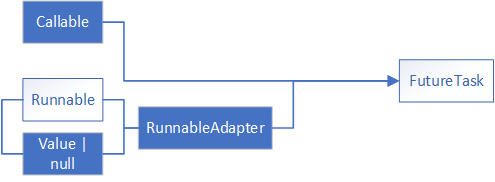

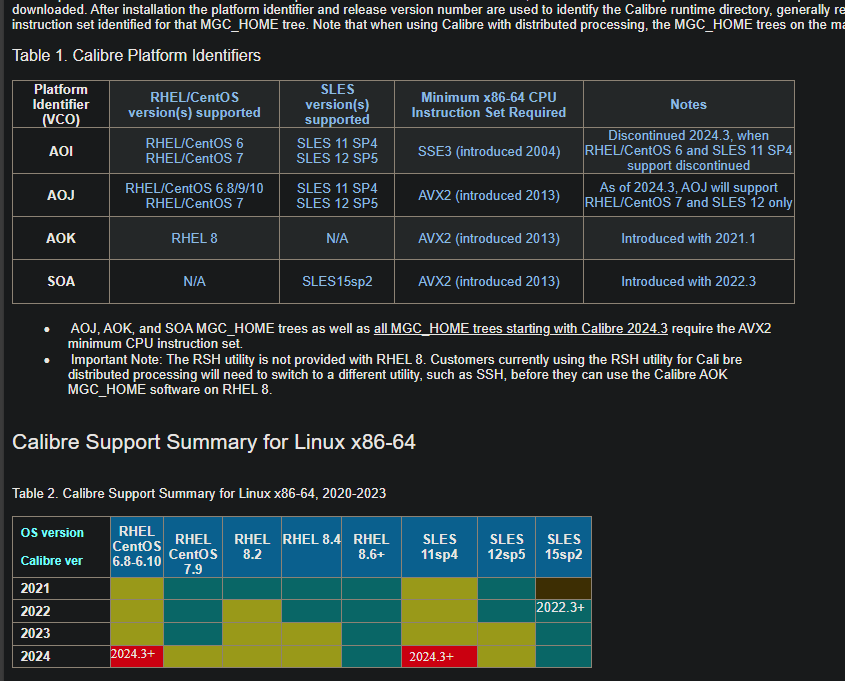
![[深入理解Java虚拟机]高效并发](./%E5%9B%BE%E7%89%87%E9%99%84%E4%BB%B6/12-2.png)