详细情况请前往
数据分析:完整的成体系的生存分析的结果汇总
科研绘图系列:组合多个文章图
介绍
本章节全面汇总了生存分析的相关各类分析方法,形成了一个系统化的生存分析教程。通过这个教程,读者可以深入了解生存分析所涵盖的多种分析技术。组合下列结果的可视化可见: 科研绘图系列:组合多个文章图
数据下载
本教程所需要的所有数据下载链接见:
- 百度网盘链接: https://pan.baidu.com/s/1KZLVvyrU1Yj78KL0CYYygQ
- 提取码:
多变量生存分析
多变量生存分析模块是一个统计分析工具,它提供了两个主要步骤来识别和评估影响生存时间的变量:
- 单变量生存分析筛选:首先,该模块基于单变量生存分析来筛选与生存时间相关的特征。这一步骤涉及对每个变量单独进行生存分析,以确定它们与生存时间的关联程度。通过这种方式,可以识别出在单变量分析中显著相关的变量,为后续的多变量分析打下基础。
- 多变量生存分析:其次,将单变量分析中显著相关的特征组合在一起,进行多变量生存分析。这一步骤旨在评估多个变量同时对生存时间的影响,以及它们之间的相互作用。多变量生存分析可以提供更全面的风险评估,因为它考虑了多个变量的联合效应,从而允许更精确地预测生存时间和识别重要的预后因素。
library(tidyverse)
library(survminer)
library(survival)
library(reshape2)
library(cowplot)
library(caret)
library(pROC)# Load data
############
load("./data/MHC_immunotherapy.RData")
ICB_data <- ICB_study[ICB_study$study == "Liu_2019", ]# Remove NA
############
ICB_data <- na.omit(ICB_data)# Binarize
#############
ICB_data$highTMB <- ICB_data$TMB > quantile(ICB_data$TMB, 0.5, na.rm = T)
ICB_data$strongMHC1 <- ICB_data$MGBS1 > quantile(ICB_data$MGBS1, 0.5, na.rm = T)
ICB_data$strongMHC2 <- ICB_data$MGBS2 > quantile(ICB_data$MGBS2, 0.5, na.rm = T)
ICB_data$strongMHCnorm <- ICB_data$MGBSnorm > quantile(ICB_data$MGBSnorm, 0.5, na.rm = T)
ICB_data$highPD1 <- ICB_data$PD1_noBatch > quantile(ICB_data$PD1_noBatch, 0.5, na.rm = T)
ICB_data$highCYT <- ICB_data$CYT_noBatch > quantile(ICB_data$CYT_noBatch, 0.5, na.rm = T)
ICB_data$highMHC1 <- ICB_data$MHC1_noBatch > quantile(ICB_data$MHC1_noBatch, 0.5, na.rm = T)
ICB_data$highMHC2 <- ICB_data$MHC2_noBatch > quantile(ICB_data$MHC2_noBatch, 0.5, na.rm = T)
ICB_data$highPurity <- ICB_data$purity > quantile(ICB_data$purity, 0.5, na.rm = T)
ICB_data$highPloidy <- ICB_data$ploidy > quantile(ICB_data$ploidy, 0.5, na.rm = T)
ICB_data$highHeterogeneity <- ICB_data$heterogeneity > quantile(ICB_data$heterogeneity, 0.5, na.rm = T)
ICB_data$highLDH <- ICB_data$LDH == 1# Save
######
dir.create("./results/data/", recursive = T)
save.image(file = "./results/data/manuscript_multivariate.RData")
saveRDS(list(Cox_model_ls = Cox_model_ls, LR_model_ls = LR_model_ls), file = "./results/data/manuscript_multivariate.rds")
相关分析
library(corrplot)
library(RColorBrewer)
library(cowplot)load("./data/MHC_immunotherapy.RData")
ICB_data <- ICB_study[ICB_study$study == "Liu_2019", ]head(ICB_data)# Create correlation plot
###########################
vars <- c("TMB", "neoAgB1", "neoAgB2", "neoAgBnorm", "pMHC1", "pMHC2", "pMHCnorm", "MGBS1", "MGBS2", "MGBSnorm", "MHC1_noBatch", "MHC2_noBatch", "heterogeneity", "ploidy", "purity", "MHC1_zyg", "MHC2_zyg", "PD1_noBatch", "B2M_noBatch", "CYT_noBatch")cor_df <- ICB_data[, vars]
colnames(cor_df) <- c("TMB", "abs. MHC-I neoAgB", "abs. MHC-II neoAgB", "abs. diff. neoAgB", "norm. MHC-I neoAgB", "norm. MHC-II neoAgB", "norm. diff. neoAgB", "MGBS-I", "MGBS-II", "MGBS-d", "MHC-I expr.", "MHC-II expr.", "heterogeneity", "ploidy", "purity", "MHC-I zygosity", "MHC-II zygosity", "PD-L1 (CD274) expr.", "B2M expr.", "CYT")# Save
#########
save.image(file = "./results/data/manuscript_correlation_analysis.RData")
saveRDS(p, file = "./results/data/manuscript_correlation_analysis.rds")
比例分析
library(survminer)
library(survival)
library(reshape2)# Load data
############
load("./data/MHC_immunotherapy.RData")ICB_data <- ICB_study[ICB_study$study == "Liu_2019", ]# MHC binders
#############
ICB_data$highTMB <- ICB_data$TMB > quantile(ICB_data$TMB, 0.5, na.rm = T)
ICB_data$strongMHC1 <- ICB_data$MGBS1 > quantile(ICB_data$MGBS1, 0.5, na.rm = T)
ICB_data$strongMHC2 <- ICB_data$MGBS2 > quantile(ICB_data$MGBS2, 0.5, na.rm = T)
ICB_data$strongMHCnorm <- ICB_data$MGBSnorm > quantile(ICB_data$MGBSnorm, 0.5, na.rm = T)# Save
######
save.image(file = "./results/data/manuscript_RECIST.RData")
saveRDS(p_BR_ls, file = "./results/data/manuscript_RECIST.rds")
生存分析
生存分析是一种统计方法,用于描述、测量和分析事件的特征,寻找其发生的原因,并对生存以及事件发生的时间进行预测。生存分析的目的在于研究哪些因素影响了事件的发生速度及生存时间的长短,它探索的因变量是一个包含了删失或者截除数据的事件时间变量。生存分析涉及的事件可以是患者的存活时间、疾病的复发、企业的存活时间等。
生存分析的目的包括但不限于以下几点:
- 描述生存时间:描述生存时间的分布情况,包括生存时间的中位数、平均值等统计量。
- 识别风险因素:识别和评估影响生存时间的各种风险因素。
- 预测生存概率:预测个体在特定时间点之前生存或事件发生的概率。
- 比较生存情况:比较不同群体或不同条件下的生存情况,例如比较不同治疗方案的效果。
- 评估干预效果:评估医疗干预、政策或其他措施对生存时间的影响。
生存分析统计方法涉及统计描述、差异性分析和回归分析三大类。其中,统计描述主要有Kaplan-Meier估计法和Life table估计法;差异性分析主要有对数秩检验和Wilcoxon test;而回归分析主要有Cox比例和非比例风险回归模型、参数回归模型。
library(survminer)
library(survival)
library(reshape2)
library(tidyverse)# Load data
############
load("./data/MHC_immunotherapy.RData")# Save
#########
save.image(file = "./results/data/manuscript_validation.RData")
saveRDS(list(p_val_ls = p_val_ls, HR_df_all = HR_df_all), file = "./results/data/manuscript_validation.rds")
生存分析2
生存分析是一种统计方法,用于描述、测量和分析事件的特征,寻找其发生的原因,并对生存以及事件发生的时间进行预测。生存分析的目的在于研究哪些因素影响了事件的发生速度及生存时间的长短,它探索的因变量是一个包含了删失或者截除数据的事件时间变量。生存分析涉及的事件可以是患者的存活时间、疾病的复发、企业的存活时间等。
生存分析的目的包括但不限于以下几点:
- 描述生存时间:描述生存时间的分布情况,包括生存时间的中位数、平均值等统计量。
- 识别风险因素:识别和评估影响生存时间的各种风险因素。
- 预测生存概率:预测个体在特定时间点之前生存或事件发生的概率。
- 比较生存情况:比较不同群体或不同条件下的生存情况,例如比较不同治疗方案的效果。
- 评估干预效果:评估医疗干预、政策或其他措施对生存时间的影响。
生存分析统计方法涉及统计描述、差异性分析和回归分析三大类。其中,统计描述主要有Kaplan-Meier估计法和Life table估计法;差异性分析主要有对数秩检验和Wilcoxon test;而回归分析主要有Cox比例和非比例风险回归模型、参数回归模型。
library(tidyverse)
library(scales)
library(survival)# Load expression data and metadata
## Load ICB metadata in an encapsulated object "all_data"
all_data <- new.env()
load("./data/MHC_immunotherapy.RData", all_data)metadata <- all_data$ICB_study# Save
#########
save.image(file = "./results/data/manuscript_validation_compare_variables.RData")
saveRDS(p_ls, file = "./results/data/manuscript_validation_compare_variables.rds")
logreg分析 & ROC分析
logreg分析和ROC分析是两种不同的统计方法,它们在机器学习和数据分析中有着各自的应用和目的。
logreg分析(Logistic Regression Analysis)
logreg分析,即逻辑回归分析,是一种用于二分类问题的统计方法。它通过估计特定变量(或变量组合)对结果发生概率的影响来工作。逻辑回归使用最大似然估计(MLE)方法来估计模型参数,即找到能够使得观测数据出现概率(似然函数)最大的参数值。
ROC分析(Receiver Operating Characteristic Analysis)
ROC分析是一种用于评估分类模型性能的方法,特别适用于比较不同分类器的性能。ROC分析通过绘制真正例率(True Positive Rate, TPR)和假正例率(False Positive Rate, FPR)的对比图来展示分类器的性能。TPR是正确分类为正例的比例,而FPR是错误分类为正例的比例。ROC曲线下的面积(AUC)是衡量分类器性能的一个重要指标,AUC值越高,表明分类器的区分能力越强
library(tidyverse)
library(survminer)
library(survival)
library(reshape2)
library(cowplot)
library(caret)
library(pROC)# Load data
load("./data/MHC_immunotherapy.RData")
p_ls <- list()# Get model from Liu - preIpi
ICB_data <- ICB_study[!is.na(ICB_study$study) & ICB_study$study == "Liu_2019", ]
ICB_data <- ICB_data[ICB_data$preIpi == T, ]ICB_data$MGBS2_z <- (mean(ICB_data$MGBS2, na.rm = T) - ICB_data$MGBS2) / sd(ICB_data$MGBS2, na.rm = T) # Z-score normalization
ICB_data$MHC2_z <- (mean(ICB_data$MHC2, na.rm = T) - ICB_data$MHC2) / sd(ICB_data$MHC2, na.rm = T) # Z-score normalization
model <- glm(R ~ MHC2_z + MGBS2_z, data = ICB_data, family = binomial)
p_ls$model <- model
# summary(model)# Save
#########
save.image(file = "./results/data/manuscript_validation_multivariate.RData")
saveRDS(p_ls, file = "./results/data/manuscript_validation_multivariate.rds")
生存分析3
生存分析是一种统计方法,用于描述、测量和分析事件的特征,寻找其发生的原因,并对生存以及事件发生的时间进行预测。生存分析的目的在于研究哪些因素影响了事件的发生速度及生存时间的长短,它探索的因变量是一个包含了删失或者截除数据的事件时间变量。生存分析涉及的事件可以是患者的存活时间、疾病的复发、企业的存活时间等。
生存分析的目的包括但不限于以下几点:
- 描述生存时间:描述生存时间的分布情况,包括生存时间的中位数、平均值等统计量。
- 识别风险因素:识别和评估影响生存时间的各种风险因素。
- 预测生存概率:预测个体在特定时间点之前生存或事件发生的概率。
- 比较生存情况:比较不同群体或不同条件下的生存情况,例如比较不同治疗方案的效果。
- 评估干预效果:评估医疗干预、政策或其他措施对生存时间的影响。
生存分析统计方法涉及统计描述、差异性分析和回归分析三大类。其中,统计描述主要有Kaplan-Meier估计法和Life table估计法;差异性分析主要有对数秩检验和Wilcoxon test;而回归分析主要有Cox比例和非比例风险回归模型、参数回归模型。
library(survminer)
library(survival)
library(reshape2)
library(tidyverse)# Load data
############
load("./data/MHC_immunotherapy.RData")
ICB_data <- ICB_study[ICB_study$study == "Liu_2019", ]
ICB_data$highTMB <- ICB_data$TMB > quantile(ICB_data$TMB, 0.5, na.rm = T)# Save
#########
save.image(file = "./results/data/manuscript_surv_neoAgB.RData")
saveRDS(list(p_TMB = p_TMB, p_pMHC1 = p_pMHC1, p_pMHC2 = p_pMHC2, p_pMHCnorm = p_pMHCnorm, p_neoAgB1 = p_neoAgB1, p_neoAgB2 = p_neoAgB2, p_neoAgBnorm = p_neoAgBnorm, fp_pMHC = fp_pMHC, fp_neoAgB = fp_neoAgB), file = "./results/data/manuscript_surv_neoAgB.rds")
生存分析4
生存分析是一种统计方法,用于描述、测量和分析事件的特征,寻找其发生的原因,并对生存以及事件发生的时间进行预测。生存分析的目的在于研究哪些因素影响了事件的发生速度及生存时间的长短,它探索的因变量是一个包含了删失或者截除数据的事件时间变量。生存分析涉及的事件可以是患者的存活时间、疾病的复发、企业的存活时间等。
生存分析的目的包括但不限于以下几点:
- 描述生存时间:描述生存时间的分布情况,包括生存时间的中位数、平均值等统计量。
- 识别风险因素:识别和评估影响生存时间的各种风险因素。
- 预测生存概率:预测个体在特定时间点之前生存或事件发生的概率。
- 比较生存情况:比较不同群体或不同条件下的生存情况,例如比较不同治疗方案的效果。
- 评估干预效果:评估医疗干预、政策或其他措施对生存时间的影响。
生存分析统计方法涉及统计描述、差异性分析和回归分析三大类。其中,统计描述主要有Kaplan-Meier估计法和Life table估计法;差异性分析主要有对数秩检验和Wilcoxon test;而回归分析主要有Cox比例和非比例风险回归模型、参数回归模型。
library(survminer)
library(survival)
library(reshape2)
library(tidyverse)# Load data
############
load("./data/MHC_immunotherapy.RData")
ICB_data <- ICB_study[ICB_study$study == "Liu_2019", ]
ICB_data$highTMB <- ICB_data$TMB > quantile(ICB_data$TMB, 0.5, na.rm = T)# Save
#########
save.image(file = "./results/data/manuscript_surv.RData")
saveRDS(p_surv_ls, file = "./results/data/manuscript_surv.rds")
差异分析
limma差异分析是一种用于基因表达数据的统计分析方法,它主要用于识别差异表达的基因。以下是对limma差异分析的详细解释:
- limma包简介: limma(Linear Models for Microarray Data)是一个R/Bioconductor软件包,它提供了一套集成的解决方案,用于分析基因表达实验数据。limma包含了丰富的功能,用于处理复杂的实验设计,并通过信息借用来克服样本量小的问题。
- 主要特点:
- 线性模型:limma使用线性模型来分析基因表达数据,这包括简单复制设计、多组实验、直接设计、因子设计和时间序列实验等。
- 经验贝叶斯统计:limma解释了经验贝叶斯测试统计量,这些统计量用于在基因表达分析中获得后验方差估计器。
- 质量权重:limma允许使用质量权重、自适应背景校正和控制点与线性模型结合使用,以提高小样本实验中基因和基因集水平的推断能力。
- 应用范围:
- 最初,limma主要用于微阵列数据的分析,但随着技术的发展,limma的能力已经显著扩展,现在可以执行RNA sequencing(RNA-seq)数据的差异表达和差异剪接分析。
- 这使得之前仅限于微阵列数据的下游分析工具现在也可以用于RNA-seq数据。
- 分析流程:
- limma分析的主要步骤包括数据预处理、线性模型拟合、经验贝叶斯方法应用以及结果的诊断和解释。
- limma提供了一系列的函数,用于存储数据或结果的不同类别,以及在线文档页面,用于每个单独的函数和每个主要步骤。
- 与其他方法的比较:
- 在RNA-seq数据的差异分析中,limma、EdgeR和DESeq2是三种有效的工具。虽然它们的分析协议在某些步骤上有所不同(例如,limma使用线性模型进行统计分析,而EdgeR和DESeq2使用负二项分布),但它们的结果部分重叠,每种方法都有其自身的优势。
library(tidyverse)
library(edgeR)
library(limma)
library(fgsea)load("./data/MHC_immunotherapy.RData")# Added: mapping table as ENSG -> HGNC must occur here (created by create_ENSG_HGNC_map_table.R)
ENSG_HGNC_map_table <- ENSG_HGNC_map_table %>%select(gene_id = ensembl_gene_id, gene = external_gene_name, gene_biotype)# Save
dir.create("./results/temp/", recursive = TRUE)
saveRDS(DGE_ls, "./results/temp/DGE_ls.rds")
功能分析
功能富集分析GSEA(Gene Set Enrichment Analysis)是一种用于解释基因表达数据的计算方法,它能够确定特定基因集是否在某种生物学状态下显著富集。GSEA的核心思想是,相比单个基因的分析,一群基因(基因集)的协同变化可能更具有生物学意义。
GSEA分析的主要步骤包括:
- 计算富集分数:评估每个基因集在排名列表中的表现,富集分数(Enrichment Score, ES)是一个介于0到1之间的值,用来衡量基因集的富集程度。
- 估计富集分数显著性水平:通过计算基因集的p值来评估其统计显著性,这通常涉及到使用排列测试来确定基因集富集分数的随机分布。
- 矫正多重假设验证:由于GSEA会同时测试多个基因集,因此需要对p值进行校正以控制假阳性率,常用的方法包括Bonferroni校正和FDR(False Discovery Rate)校正。
GSEA分析的目的在于:
- 识别生物过程中的关键基因集:GSEA能够识别在特定条件下显著富集的基因集,这些基因集可能涉及特定的生物学过程或疾病相关的通路。
- 揭示基因表达的总体趋势:GSEA不仅关注差异表达的基因,而是利用所有基因的表达数据,揭示整个基因集的表达趋势,从而判断特定通路是被激活还是抑制。
- 提高数据分析的可解释性:通过将基因集与已知的生物学知识相联系,GSEA提高了基因表达数据的解释性,使得研究者能够更好地理解数据背后的生物学意义。
library(tidyverse)
library(ggrepel)
library(fgsea)
library(cowplot)load("./data/MHC_immunotherapy.RData")
DGE_ls <- readRDS("./results/temp/DGE_ls.rds")# Save
#########
p_DGE_ls <- list(p_fGSEA = fGSEA_plot, p_RS_ls = p_RS_ls, p_GSEA_preIpi = p_GSEA_preIpi)
save.image(file = "./results/data/manuscript_DGE_analysis.RData")
saveRDS(p_DGE_ls, file = "./results/data/manuscript_DGE_analysis.rds")
介绍
组合多个文章图,图所需要的结果文件见:数据分析:完整的成体系的生存分析的结果汇总
数据下载
数据下载链接见:
- 百度网盘链接: https://pan.baidu.com/s/1KZLVvyrU1Yj78KL0CYYygQ
- 提取码:
图所需要的结果文件见:数据分析:完整的成体系的生存分析的结果汇总
图1
p_ls <- readRDS("./results/data/manuscript_surv.rds")
p_BR_ls <- readRDS("./results/data/manuscript_RECIST.rds")# Create fig
p1 <- p_ls$p_TMB +ggtitle("Tumor Mutation Burden")
p2 <- p_ls$p_strongMHC1 +ggtitle("MGBS-I")
p3 <- p_ls$p_strongMHC2 +ggtitle("MGBS-II")
p4 <- p_ls$p_strongMHCnorm +ggtitle("MGBS-d")dir.create("./results/figs/", recursive = T)
ggplot2::ggsave(paste0("./results/figs/manuscript_fig1.pdf"), p, width = 178, height = 265 / 1.5, units = "mm")
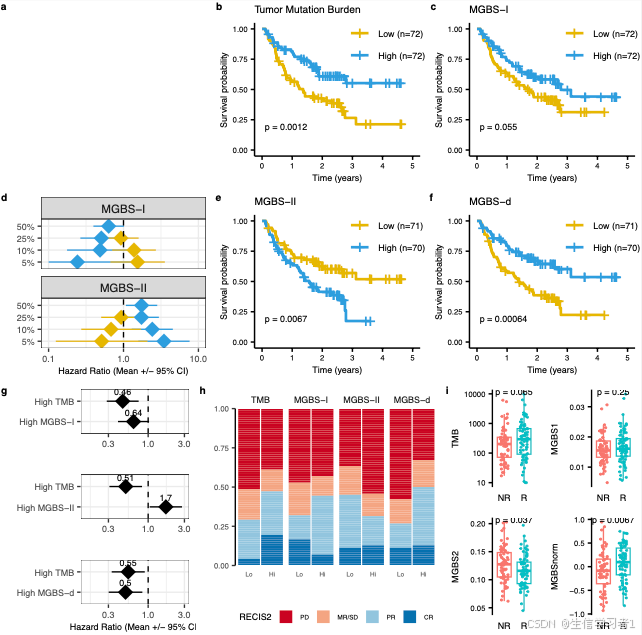
图2
p_ls <- readRDS("./results/data/manuscript_surv.rds")p1 <- plot_grid(NA, p_ls$p_ipi_vs_noIpi,ncol = 2
)ggplot2::ggsave(paste0("./results/figs/manuscript_fig2.pdf"), p, width = 178, height = 0.5 * 265, units = "mm")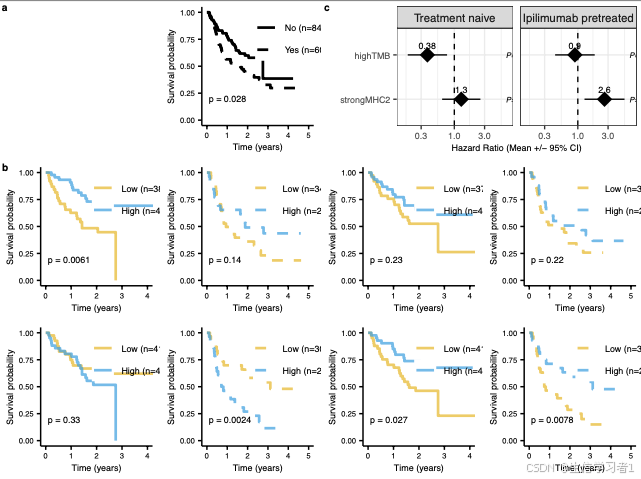
图3
p_ls <- readRDS("./results/data/manuscript_multivariate.rds")# ROC & surv
p_ROC_surv <- plot_grid(p_ls$LR_model_ls$"TRUE"$p_ROC, p_ls$LR_model_ls$"TRUE"$p_surv, NA,ncol = 1,labels = c("a", "c"),label_size = 8
)ggplot2::ggsave("./results/figs/manuscript_fig3.pdf", p, width = 178, height = 265 / 2, units = "mm")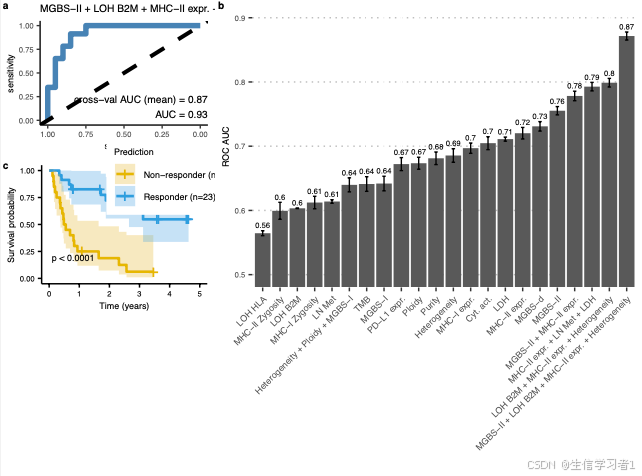
图4
p_ls <- readRDS("./results/data/manuscript_DGE_analysis.rds")ggplot2::ggsave("./results/figs/manuscript_fig4.pdf", p, width = 3 / 4 * 178, height = 265 / 3, units = "mm")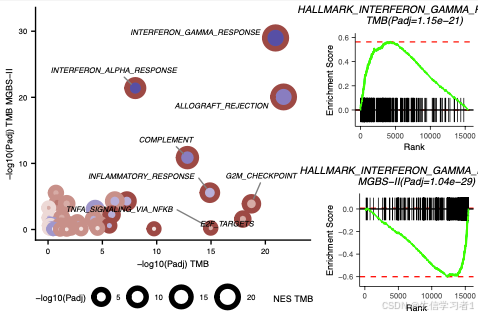
附图2
p_ls <- readRDS("./results/data/manuscript_surv.rds")p <- plot_grid(p_ls$p_th_ls$TMBclass$`0.75` + ggtitle("Tumor Mutation Burden") + theme(legend.background = element_blank()),p_ls$p_th_ls$TMBclass$`0.9` + ggtitle("Tumor Mutation Burden") + theme(legend.background = element_blank()),p_ls$p_th_ls$TMBclass$`0.95` + ggtitle("Tumor Mutation Burden") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHC1class$`0.75` + ggtitle("MGBS-I") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHC1class$`0.9` + ggtitle("MGBS-I") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHC1class$`0.95` + ggtitle("MGBS-I") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHC2class$`0.75` + ggtitle("MGBS-II") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHC2class$`0.9` + ggtitle("MGBS-II") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHC2class$`0.95` + ggtitle("MGBS-II") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHCnormclass$`0.75` + ggtitle("MGBS-d") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHCnormclass$`0.9` + ggtitle("MGBS-d") + theme(legend.background = element_blank()),p_ls$p_th_ls$MHCnormclass$`0.95` + ggtitle("MGBS-d") + theme(legend.background = element_blank()),ncol = 3
)ggplot2::ggsave(paste0("./results/figs/manuscript_figS2_surv_th.pdf"), p, width = 178, height = 265, units = "mm")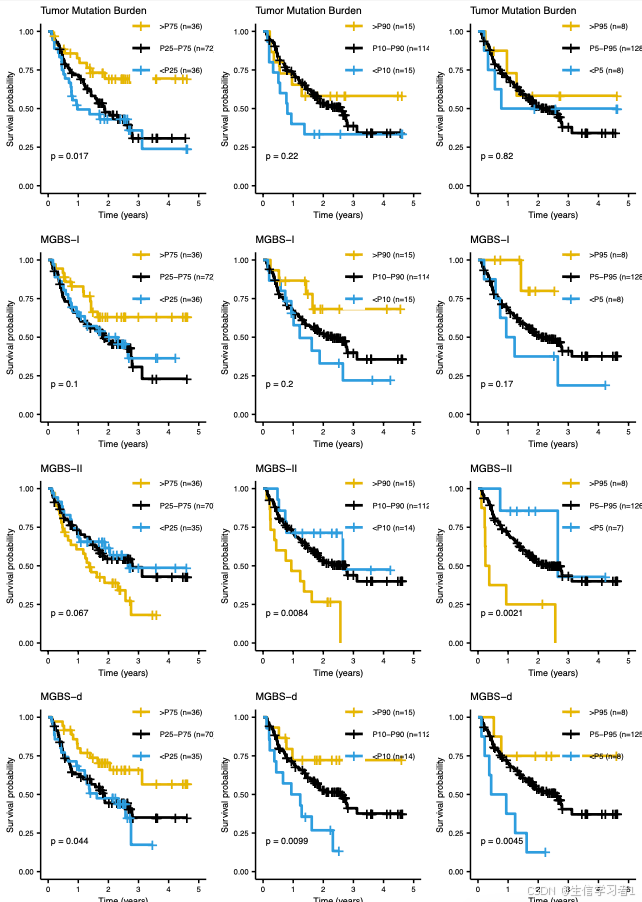
附图3
p <- readRDS("./results/data/manuscript_correlation_analysis.rds")# Create figure
ggplot2::ggsave(paste0("./results/figs/manuscript_figS3_correlation.pdf"), p, width = 178, height = 265, units = "mm")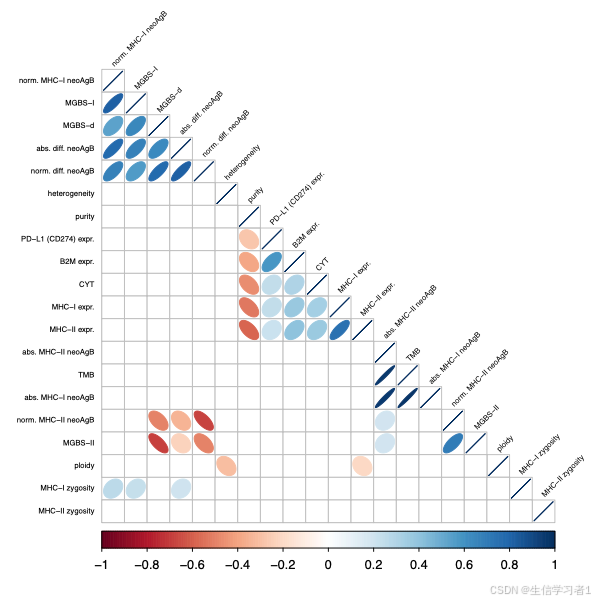
附图4
p_ls <- readRDS("./results/data/manuscript_validation.rds")# Forest plot
# Adjust sizes facet
gt <- ggplot_gtable(ggplot_build(p_ls$p_val_ls$fp))
# gtable_show_layout(gt)
gt$heights[10] <- 1 / 2 * gt$heights[10]
# fp<- grid.draw(gt)# Forest plot preIpi
fp_preIpi <- p_ls$p_val_ls$fp_preIpi# Create figure
ggplot2::ggsave(paste0("./results/figs/manuscript_figS4_validation.pdf"), p, width = 178, height = 265, units = "mm")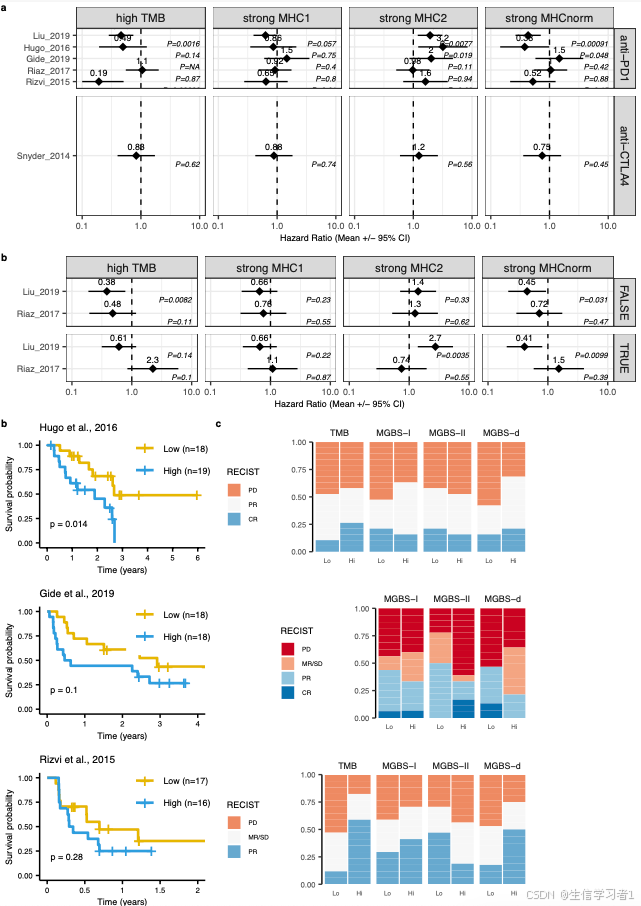
附图5
p_ls <- readRDS("./results/data/manuscript_surv_neoAgB.rds")p <- plot_grid(p_ls$fp_neoAgB + ggtitle("Absolute Neoantigen Burden") + theme(title = element_text(size = 8)), p_ls$fp_pMHC + ggtitle("Normalized Neoantigen Burden") + theme(title = element_text(size = 8)),p_ls$p_neoAgB1 + ggtitle("MHC-I NeoAgB"), p_ls$p_pMHC1 + ggtitle("norm. MHC-I NeoAgB"),p_ls$p_neoAgB2 + ggtitle("MHC-II NeoAgB"), p_ls$p_pMHC2 + ggtitle("norm. MHC-II NeoAgB"),p_ls$p_neoAgBnorm + ggtitle("diff. NeoAgB"), p_ls$p_pMHCnorm + ggtitle("norm. diff. NeoAgB"),ncol = 2
)# Create figures
ggplot2::ggsave(paste0("./results/figs/manuscript_figS5_neoAgB.pdf"), p_neoAgB, width = 178, height = 265, units = "mm")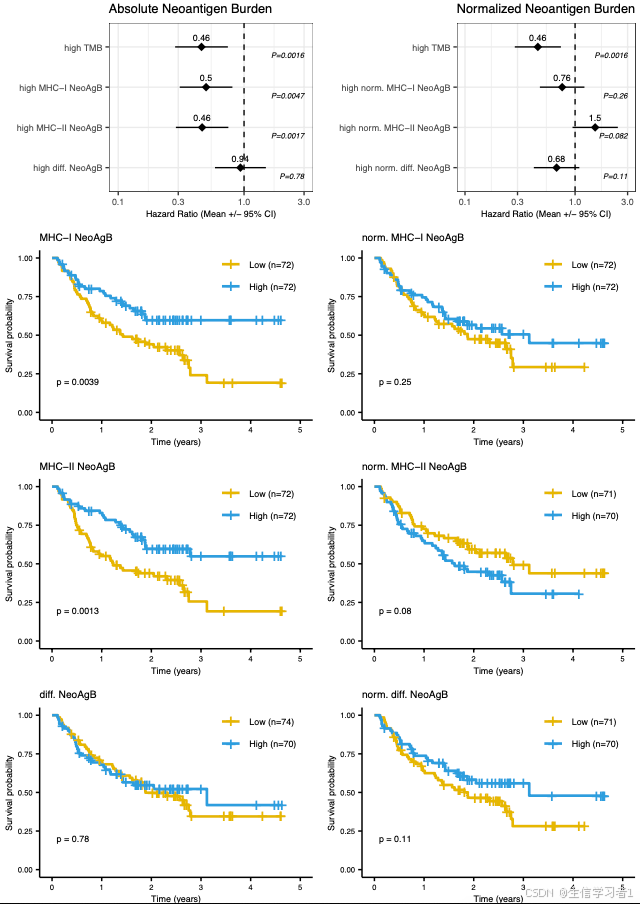
附图6-7
p_ls <- readRDS("./results/data/manuscript_multivariate.rds")# 1. COX
t_preIpi <- p_ls$Cox_model_ls$"TRUE"$t_uni
t_ipiNaive <- p_ls$Cox_model_ls$"FALSE"$t_unip1 <- plot_grid(t_ipiNaive, t_preIpi,ncol = 2
)p_cox <- plot_grid(p1, p_ls$Cox_model_ls$fp_multi + scale_y_continuous(limits = c(0.15, 12), trans = "log10"), NA,ncol = 1,rel_heights = c(1, 1 / 2, 1)
)# Create figures
ggplot2::ggsave(paste0("./results/figs/manuscript_figS6_LogReg.pdf"), p_logReg, width = 178, height = 265, units = "mm")
ggplot2::ggsave(paste0("./results/figs/manuscript_figS7_Cox.pdf"), p_cox, width = 178, height = 265, units = "mm")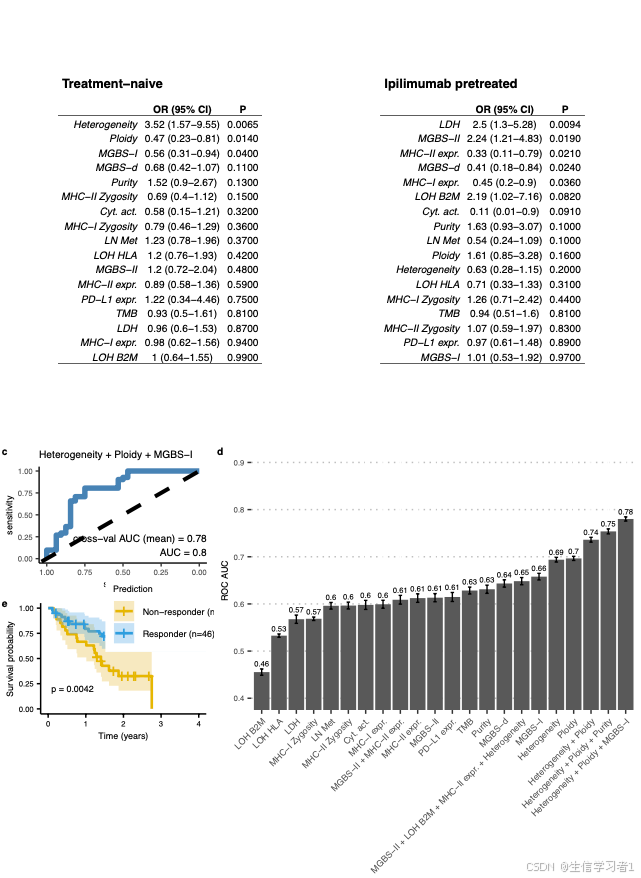
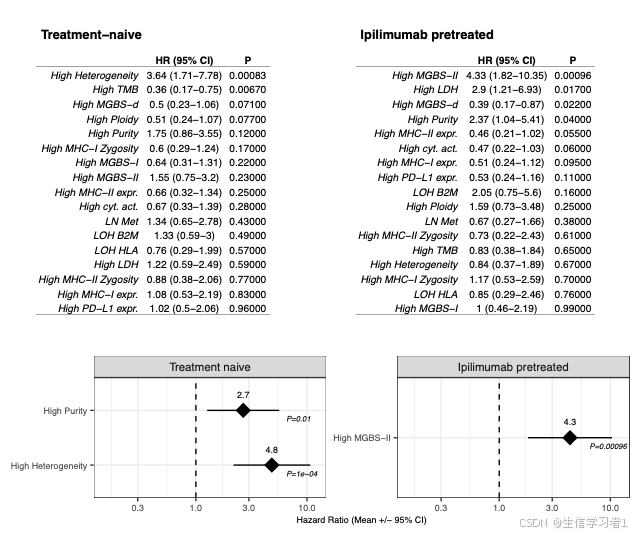
附图8
p_ls <- readRDS("./results/data/manuscript_validation_multivariate.rds")
p_compare_ls <- readRDS("./results/data/manuscript_validation_compare_variables.rds")# Create figure
ggplot2::ggsave(paste0("./results/figs/manuscript_figS8_validation_multivariate.pdf"), p, width = 178, height = 265, units = "mm")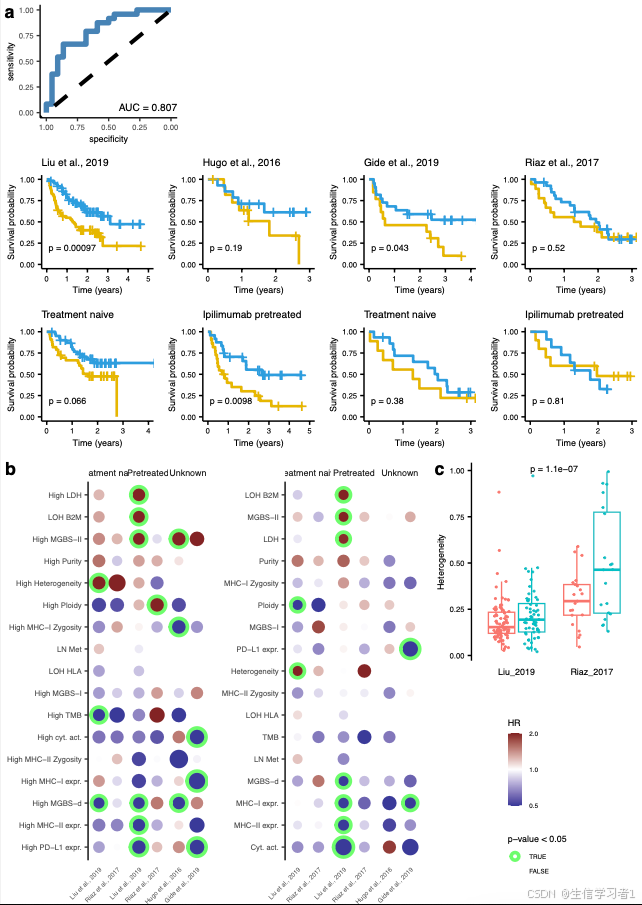
附图9
p_ls <- readRDS("./results/data/manuscript_DGE_analysis.rds")ggplot2::ggsave(paste0("./results/figs/manuscript_figS9_GSEA.pdf"), p, width = 178, height = 265, units = "mm")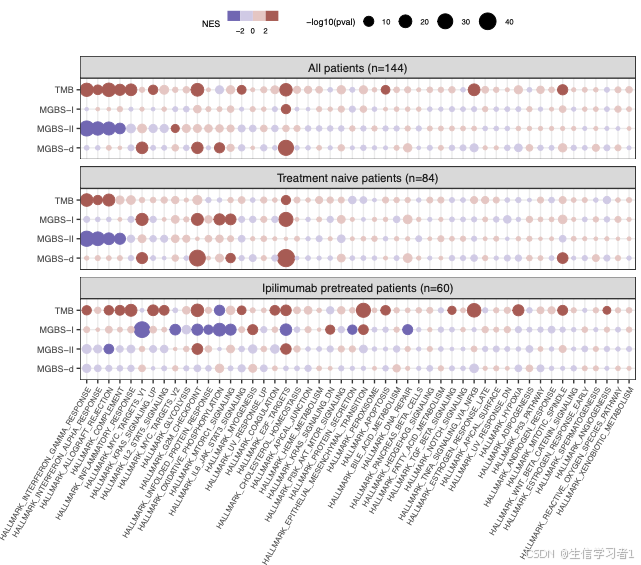
系统信息
- 数据分析
R version 4.3.3 (2024-02-29)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.2Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8time zone: Asia/Shanghai
tzcode source: internalattached base packages:
[1] grid stats graphics grDevices utils datasets methods base other attached packages:[1] lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2 readr_2.1.5 [7] tidyr_1.3.1 tibble_3.2.1 tidyverse_2.0.0 gridExtra_2.3 ggplot2_3.5.1 cowplot_1.1.3 loaded via a namespace (and not attached):[1] vctrs_0.6.5 cli_3.6.3 rlang_1.1.4 stringi_1.8.3 generics_0.1.3 [6] glue_1.7.0 colorspace_2.1-0 hms_1.1.3 scales_1.3.0 fansi_1.0.6
[11] munsell_0.5.0 tzdb_0.4.0 lifecycle_1.0.4 compiler_4.3.3 timechange_0.3.0
[16] pkgconfig_2.0.3 rstudioapi_0.16.0 R6_2.5.1 tidyselect_1.2.1 utf8_1.2.4
[21] pillar_1.9.0 magrittr_2.0.3 tools_4.3.3 withr_3.0.0 gtable_0.3.4
- 画图
R version 4.3.3 (2024-02-29)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.2Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8time zone: Asia/Shanghai
tzcode source: internalattached base packages:
[1] grid stats graphics grDevices utils datasets methods base other attached packages:[1] ggrepel_0.9.5 fgsea_1.28.0 edgeR_4.0.16 limma_3.58.1 scales_1.3.0 pROC_1.18.5 [7] caret_6.0-94 lattice_0.22-6 reshape2_1.4.4 survival_3.7-0 survminer_0.4.9 ggpubr_0.6.0
[13] lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2 readr_2.1.5
[19] tidyr_1.3.1 tibble_3.2.1 tidyverse_2.0.0 gridExtra_2.3 ggplot2_3.5.1 cowplot_1.1.3 loaded via a namespace (and not attached):[1] tidyselect_1.2.1 timeDate_4032.109 digest_0.6.35 rpart_4.1.23 [5] timechange_0.3.0 lifecycle_1.0.4 statmod_1.5.0 magrittr_2.0.3 [9] compiler_4.3.3 rlang_1.1.4 tools_4.3.3 utf8_1.2.4
[13] data.table_1.15.2 knitr_1.45 ggsignif_0.6.4 plyr_1.8.9
[17] BiocParallel_1.36.0 abind_1.4-5 withr_3.0.0 nnet_7.3-19
[21] stats4_4.3.3 fansi_1.0.6 xtable_1.8-4 colorspace_2.1-0
[25] future_1.33.1 globals_0.16.3 iterators_1.0.14 MASS_7.3-60.0.1
[29] cli_3.6.3 generics_0.1.3 rstudioapi_0.16.0 future.apply_1.11.1
[33] km.ci_0.5-6 tzdb_0.4.0 splines_4.3.3 parallel_4.3.3
[37] survMisc_0.5.6 vctrs_0.6.5 hardhat_1.3.1 Matrix_1.6-5
[41] carData_3.0-5 car_3.1-2 hms_1.1.3 rstatix_0.7.2
[45] listenv_0.9.1 locfit_1.5-9.9 foreach_1.5.2 gower_1.0.1
[49] recipes_1.0.10 glue_1.7.0 parallelly_1.37.1 codetools_0.2-19
[53] stringi_1.8.3 gtable_0.3.4 munsell_0.5.0 pillar_1.9.0
[57] ipred_0.9-14 lava_1.8.0 R6_2.5.1 KMsurv_0.1-5
[61] backports_1.4.1 broom_1.0.5 class_7.3-22 fastmatch_1.1-4
[65] Rcpp_1.0.12 nlme_3.1-164 prodlim_2023.08.28 xfun_0.43
[69] zoo_1.8-12 pkgconfig_2.0.3 ModelMetrics_1.2.2.2