前面五节,我们使用阿尔兹海默症数据做了一个数据预处理案例,包括如下内容:
GEO生信数据挖掘(一)数据集下载和初步观察
GEO生信数据挖掘(二)下载基因芯片平台文件及注释
GEO生信数据挖掘(三)芯片探针ID与基因名映射处理
GEO生信数据挖掘(四)数据清洗(离群值处理、低表达基因、归一化、log2处理)
GEO生信数据挖掘(五)提取临床信息构建分组,分组数据可视化(绘制层次聚类图,绘制PCA图)
本节目录
结核病基因表达数据(GSE107994)观察
临床形状数据预处理
基因表达数据预处理
绘图观察数据
结核病基因表达数据(GSE107994)观察
由于,在数据分析过程,你拿的数据样式可能会有不同,本节我们以结核病基因表达数据(GSE107994)为例,做一个实践案例。该数据集的临床形状数据和基因表达数据是单独分开的,读取,和处理都需自己改动代码。

先来看看基因表达数据,这个探针注释工作已经完成了,不需要处理。

再看看临床形状数据,需要手工删除前面的注释,把后半部分规整的数据保留下来。

临床形状数据预处理
# 手工删除前面的注释,读文件,转置
pdata <- t(read.delim("GSE107994_series_matrix_clean.txt", header = TRUE, sep = "\t"))
# 手工删除前面的注释,读文件,转置
pdata <- t(read.delim("GSE107994_series_matrix_clean.txt", header = TRUE, sep = "\t"))
pdata <-pdata[-1,]
pdata_info = pdata[,c(1,7)]
colnames(pdata_info) = c('geo_accession','type')#观察样本类型的取值都有哪些(结核,潜隐进展,对照和潜隐)
unique(pdata_info[,2])
#"Leicester_Active_TB" "Longitudnal_Leicester_LTBI_Progressor" "Leicester_Control" #"Leicester_LTBI" group_data = as.data.frame(pdata_info)处理前
处理后
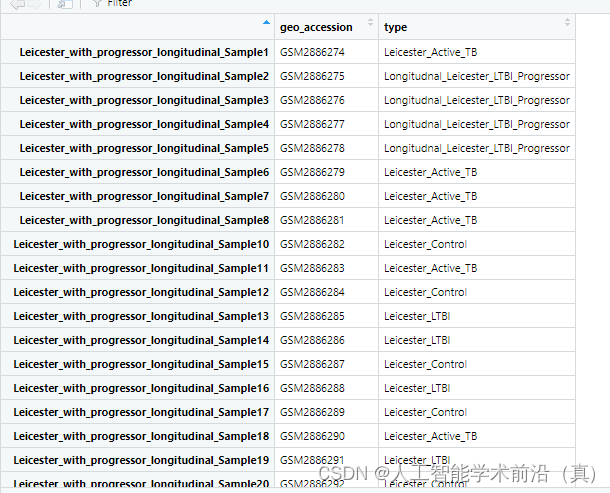
增加不同的类型标签,根据需要,选取实验组和对照组
# 使用grepl函数判断字符串是否包含'Control',并进行相应的修改
group_data$group_easy <- ifelse(grepl("Control", group_data$type ), "Control", "TB")# 使用grepl函数判断字符串是否包含特定内容,然后进行相应的修改
group_data$group_easy <- ifelse(grepl("Control", group_data$type), "Control",ifelse(grepl("LTBI", group_data$type), "LTBI","TB"))
# 使用grepl函数判断字符串是否包含特定内容,然后进行相应的修改
group_data$group_more <- ifelse(grepl("Control", group_data$type), "Control",ifelse(grepl("LTBI_Progressor", group_data$type), "LTBI_Progressor",ifelse(grepl("LTBI", group_data$type), "LTBI","TB")))#尝试把进展组排除出去save(group_data,file = "group_data.Rdata")
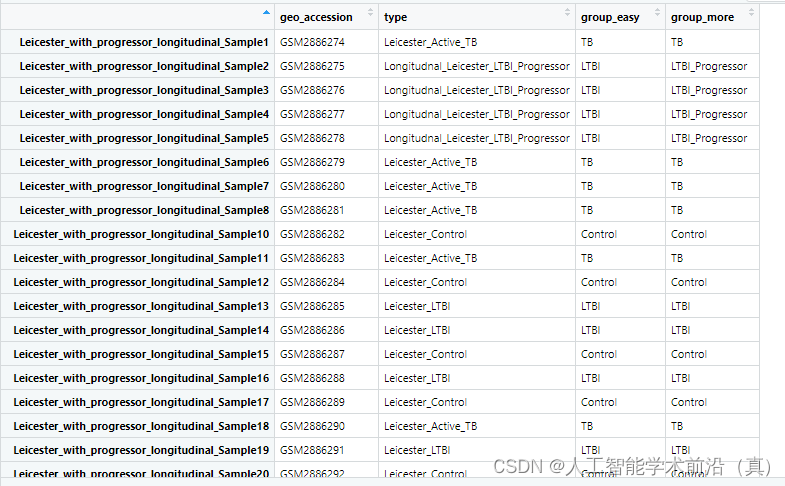
例如 我们可以进行 TB(结核) 和LTBI(潜隐结核)实验对照分析。
基因表达数据预处理
读取数据集
install.packages("openxlsx")
library(openxlsx)# 读基因表达矩阵,第一列为基因名ID
gse_info<- as.data.frame(read.xlsx("GSE107994_Raw.xlsx", sheet = 1))
colnames(gse_info)
后续运行代码过程中,发现基因名称中有全数字的情况,这里做删除操作。
library(dplyr)
dim(gse_info)
#基因里面有数字
gse_info <- gse_info[!grepl("^\\d+$", gse_info$ID), ] #有效#基因名全为空
gse_info = gse_info[gse_info$ID != "",] #无剔除
dim(gse_info) #[1] 58023 176#负值处理
gse_info[gse_info <= 0] <- 0.0001#重复值检查
table(duplicated(gse_info$ID))分组数据条件筛选,TB(结核) 和LTBI(潜隐结核)
#+=====================================================
#================================================
#+========type分组数据条件筛选step3===========
#+====================================#预处理之前,先筛选出TB组和LTBI组 的数据
unique(group_data[,"group_more"]) #"TB" "LTBI_Progressor" "Control" "LTBI" #"TB" "LTBI" 对照,则剔除 "LTBI_Progressor" "Control"
geo_accession_TB_LTBI <- group_data[group_data$group_more == "LTBI_Progressor" | group_data$group_more == "Control","geo_accession"]
gse_TB_FTBI = gse_info[,!(names(gse_info) %in% geo_accession_TB_LTBI)]gse_TB_FTBI
低表达过滤(平均值小于1)
#+=====================================================
#================================================
#+========删除 低表达(平均值小于1)基因 step4===========
#+====================================
#+==============================#新增一列计算平均
gene_avg_expression <- rowMeans(gse_TB_FTBI[, -1]) # 计算每个基因的平均表达量,排除第一列(基因名)
#仅去除在所有样本里表达量都为零的基因(平均值小于1)
gse_TB_FTBI_filtered_genes_1 <- gse_TB_FTBI[gene_avg_expression >= 1, ]低表达过滤方案二(保留样本表达的排名前50%的基因)
#+=================================================================
#============================================================
#+========删除 低表达(排名前50%)基因 step5===========
#+==========================================
#+================================#仅保留在一半以上样本里表达的基因# 计算基因表达矩阵每个基因的平均值
gene_means <- rowMeans(gse_TB_FTBI_filtered_genes_1[,-1])# 计算基因平均值的排序百分位数
gene_percentiles <- rank(gene_means) / length(gene_means)# 获取阈值
threshold <- 0.25 # 删除后25%的阈值
#threshold <- 0.5 # 删除后50%的阈值
# 根据阈值筛选低表达基因
gse_TB_FTBI_filtered_genes_2 <- gse_TB_FTBI_filtered_genes_1[gene_percentiles > threshold, ]# 打印筛选后的基因表达矩阵
dim(gse_TB_FTBI_filtered_genes_2) #[1] 17049 176
删除重复基因,取平均
#+=================================================================
#============================================================
#+========重复基因,取平均值 step6===========
#+==========================================
#+================================dim(filtered_genes_2)
table(duplicated(filtered_genes_2$ID))#把重复的Symbol取平均值
averaged_data <- aggregate(.~ID , filtered_genes_2, mean, na.action = na.pass) ##把重复的Symbol取平均值#把行名命名为SYMBOL
row.names(averaged_data) <- averaged_data$ID
dim(averaged_data)#去掉缺失值
matrix_na = na.omit(averaged_data) #删除Symbol列(一般是第一列)
matrix_final <- subset(matrix_na, select = -1)
dim(matrix_final) #[1] 22687 175离群值处理
#+=================================================================
#============================================================
#+========离群值处理 step7==========================
#+==========================================
#+================================#数据离群处理
#处理极端值
#定义向量极端值处理函数
#用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
dljdz=function(x) {DOWNB=quantile(x,0.25)-1.5*(quantile(x,0.75)-quantile(x,0.25))UPB=quantile(x,0.75)+1.5*(quantile(x,0.75)-quantile(x,0.25))x[which(x<DOWNB)]=quantile(x,0.5)x[which(x>UPB)]=quantile(x,0.5)return(x)
}#第一列设置为行名
matrix_leave=matrix_final_TB_LTBIboxplot(matrix_leave,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave)#处理离群值
matrix_leave_res=apply(matrix_leave,2,dljdz)boxplot(matrix_leave_res,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave_res)log2 处理
#+=================================================================
#============================================================
#+========log2 处理 step8==========================
#+==========================================
#+================================# limma的函数归一化,矫正差异 ,表达矩阵自动log2化#1.归一化不是绝对必要的,但是推荐进行归一化。
#有重复的样本中,应该不具备生物学意义的外部因素会影响单个样品的表达,
#例如中第一批制备的样品会总体上表达高于第二批制备的样品,假设所有样品表达值的范围和分布都应当相似,
#需要进行归一化来确保整个实验中每个样本的表达分布都相似。
#2.归一化要在log2标准化之前做library(limma) exprSet=normalizeBetweenArrays(matrix_leave_res)boxplot(exprSet,outline=FALSE, notch=T, las=2) ##出箱线图## 这步把矩阵转换为数据框很重要
class(exprSet) ##注释:此时数据的格式是矩阵(Matrix)
exprSet <- as.data.frame(exprSet)#标准化 表达矩阵自动log2化
qx <- as.numeric(quantile(exprSet, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||(qx[6]-qx[1] > 50 && qx[2] > 0) ||(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)#负值全部置为空
#exprSet[exprSet <= 0] <- 0.0001
#去掉缺失值
#exprSet = na.omit(exprSet) #15654
#save (exprSet,file = "waitlog_data_TB_LTBI.Rdata")## 开始判断
if (LogC) { exprSet [which(exprSet <= 0)] <- NaN## 取log2exprSet_clean <- log2(exprSet+1) #@@@@是否加一 加一的话不产生负值@#@¥@#@#@%@%¥@@@@@@print("log2 transform finished")
}else{print("log2 transform not needed")
}boxplot(exprSet_clean,outline=FALSE, notch=T, las=2) ##出箱线图dataset_TB_LTBI =exprSet_clean绘图观察数据
#+=================================================================
#============================================================
#+========对照组不同颜色画箱线图 step9==========================
#+==========================================
#+================================# 使用grepl函数判断字符串是否包含'LTBI',并进行颜色标记,为了画图
group_data_TB_LTBI$group_color <- ifelse(grepl("LTBI", group_data_TB_LTBI$group_more), "yellow", "blue")#画箱线图查看数据分布
group_list_color = group_data_TB_LTBI$group_color
boxplot( data.frame(dataset_TB_LTBI),outline=FALSE,notch=T,col=group_list_color,las=2)dev.off()#+=================================================================
#============================================================
#+========绘制层次聚类图 step10==========================
#+==========================================
#+================================
#+#检查表达矩阵的样本名称,和分租信息的样本名称顺序,是否一致对应
colnames(dataset_TB_LTBI)
group_data_TB_LTBI$geo_accessionexprSet =dataset_TB_LTBI
#修改GSM的名字,改为分组信息
#colnames(exprSet)=paste(group_list,1:ncol(exprSet),sep = '')#定义nodePar
nodePar=list(lab.cex=0.6,pch=c(NA,19),cex=0.7,col='blue')
#聚类
hc=hclust(dist(t(exprSet))) #t()的意思是转置#绘图
plot(as.dendrogram(hc),nodePar = nodePar,horiz = TRUE)dev.off()#+=================================================================
#============================================================
#+========绘制PCA散点样本可视化图 step11===================
#+==========================================
#+================================##PCA图
#install.packages('ggfortify')
library(ggfortify)
df=as.data.frame(t(exprSet)) #转置后就变成了矩阵
dim(df) #查看数据维度
dim(exprSet)df$group=group_data_TB_LTBI$group_more #加入样本分组信息
autoplot(prcomp(df[,1:ncol(df)-1]),data=df,colour='group') #PCA散点图dev.off()至此,我们对两个数据集进行了预处理工作,下面我们可以对处理完毕的数据进行差异分析了。