VASP软件是基于贋势和平面波基组的第一性原理密度泛函计算程序。VASP使用的是平面波基组,电子与原子核之间的相互作用使用投影缀加波贋势(Projector Augmented Wave,PAW)方法描述,从而进行量子力学计算。VASP采用PAW贋势,使得平面波基组尺寸非常小,描述原子一般不超过100个平面波基组,大多数情况下每原子50个平面波就能得到可靠结果。

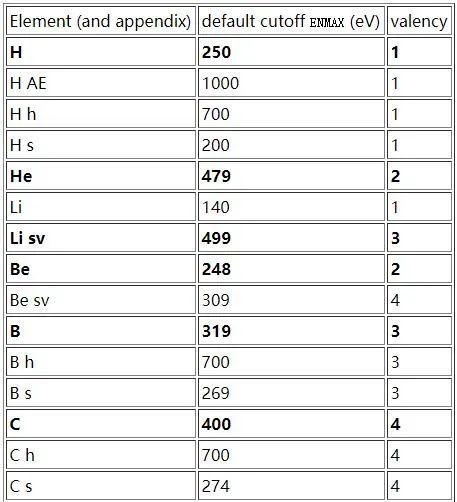
VASP赝势按产生方法不同可分为USPP(ultrasoftpesudopotential)和PAW(projector augmented wave method),两种方法都可以相当程度地减少过渡金属或第一行元素的每个原子所必需的平面波数量。按交换关联函数的不同又可以有 LDA (local density approximation) 和 GGA(generalized gradient approximation),其中GGA之下又可以再分为PW91和PBE。按原子中是否处理的半芯态(semi-core),分为A,A_sv,A_pv和A_d。按赝势文件中截断能参数ENMAX的大小,分为普通赝势A,软赝势A_s(精度较低,速度较快)和硬赝势A_h(精度较高,速度较慢)。USPP型赝势所需截断能较小,计算速度快,PAW赝势通常较大的截断能,而且考虑的电子数多,计算慢,但精确度高。对于化合物(不同原子半径的元素混合)来说,PAW赝势比超软赝势精确度高。
所以,在选择赝势的时候一定要考虑全面,根据体系的特性以及所含元素种类选择合适的赝势。VASP官网推荐了一些赝势,笔者仅截取了一部分,其中加粗显示的即为推荐的,完整版点击文末链接访问VASP官网。在侯柱峰老师的个人经验手册里面对于选择赝势给出了很详细的建议,这里引用侯柱峰老师手册的一段话:“在处理磁性材料,所计算的体系含有碱金属、碱土金属、周期表左边的3d 过渡元素、镧系和锕系元素时,强烈推荐用PAW 势,计算精度有提高。在采用超越赝势(USPP)时,使用PW91 的GGA 时,强烈要求把VOSKOWN = 1 给选上。在采用PAW 势时,一般推荐用LDA 和PBE 的。”所以,各位朋友在用VASP做计算时,选取赝势一定要先阅读VASP官网,查找相应文献。
对于生成POTCAR文件的方法,在前面教程已经给出了,即“cat/…/PAW_PBE/{A1,A2,A3,…,An}/POTCAR >> POTCAR”,其中/…/表示赝势包的路径,PAW_PBE代表赝势种类,{A1,A2,A3,…,An}代表原子种类。一定要注意,要按照POSCAR 文件各类原子的顺序,依次使用上面的命令。同时推荐大家使用大师兄所写的脚本来产生POTCAR,大师兄的脚本可以更加方便的产生POTCAR文件,脚本请查阅大师兄科研网。
参考资料:
源资官网:http://www.tri-ibiotech.com.cn/vasp/product_17.html
VASP官方推荐赝势:https://cms.mpi.univie.ac.at/vasp/vasp/Recommended_PAW_potentials_DFT_calculations_using_vasp_5_2.html
VASP问世25年来,累计发表SCI论文超过90000篇,其中大量是实验与计算相结合的文章。其计算结果不仅可以验证实验结果,更能够预测实验结果甚至设计实验。
实验与计算已成为顶刊标配,对计算感兴趣可以后台私信“计算”获取相关资料!