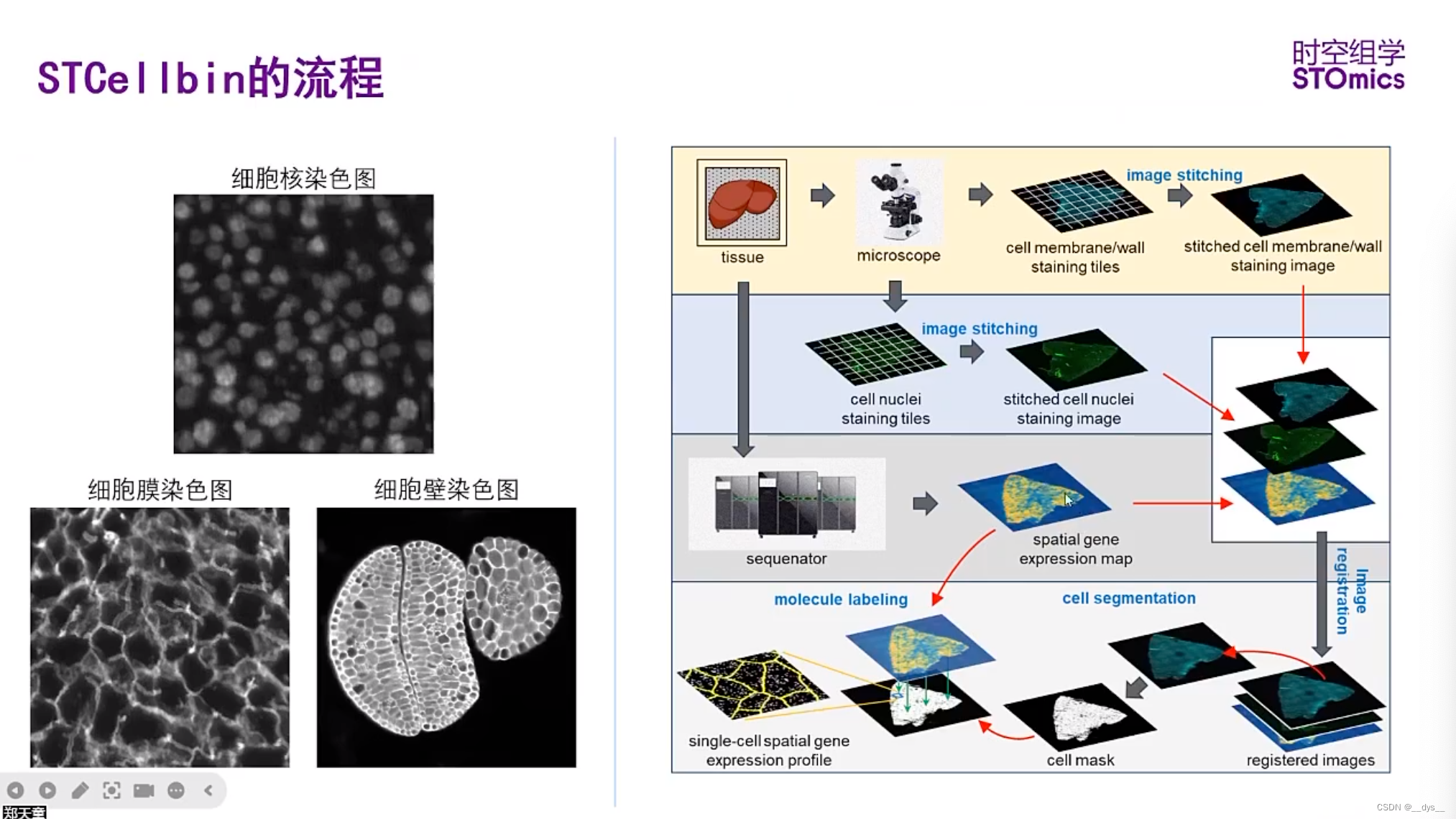
简介
STCellbin 利用细胞核染色图像作为桥梁来获取与空间基因表达图谱对齐的细胞膜/壁染色图像。通过采用先进的细胞分割技术,可以获得准确的细胞边界,从而获得更可靠的单细胞空间基因表达谱。此次更新的增强功能为细胞内基因表达的空间组织提供了宝贵的见解,并有助于更深入地了解组织生物学。
安装
CPU版
使用mamba安装虚拟环境和依赖包【mamba安装可以查看:Mamba OuttttttStanding (qq.com)】。当前版本适用于python3.8
git clone https://github.com/STOmics/STCellbin.git
mamba create --name=STCellbin python=3.8
conda activate STCellbin
mamba install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 -c pytorch
cd STCellbin
pip install -r requirements.txt -i https://pypi.tuna.tsinghua.edu.cn/simple
GPU版
三步走:CUDA版本-Pytroch版本-Python版本
-
查看GPU版本号:
nvidia-smi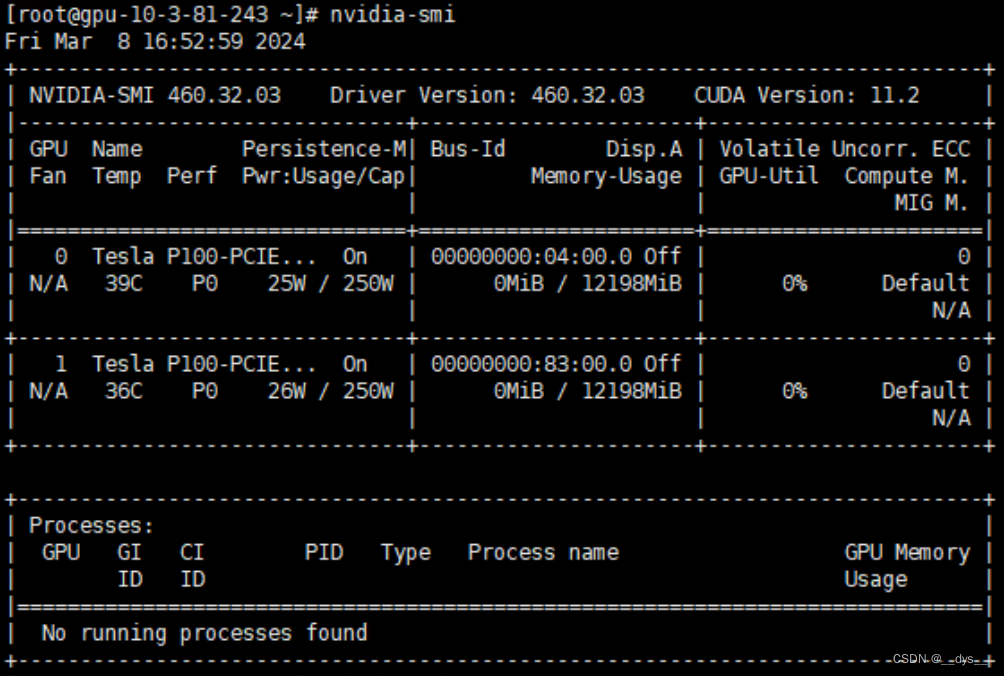
-
查看CUDA11.2使用的Pytroch版本号Previous PyTorch Versions | PyTorch
## 没有找到CUDA 11.2对应版本,选择11.3。CUDA 11.3对应的pytroch是1.12
# CUDA 10.2
conda install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 cudatoolkit=10.2 -c pytorch
# CUDA 11.3
conda install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 cudatoolkit=11.3 -c pytorch
# CUDA 11.6
conda install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 cudatoolkit=11.6 -c pytorch -c conda-forge
# CPU Only
conda install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 cpuonly -c pytorch
- 查看pytroch1.12适配的python版本:pytorch/vision: Datasets, Transforms and Models specific to Computer Vision (github.com)
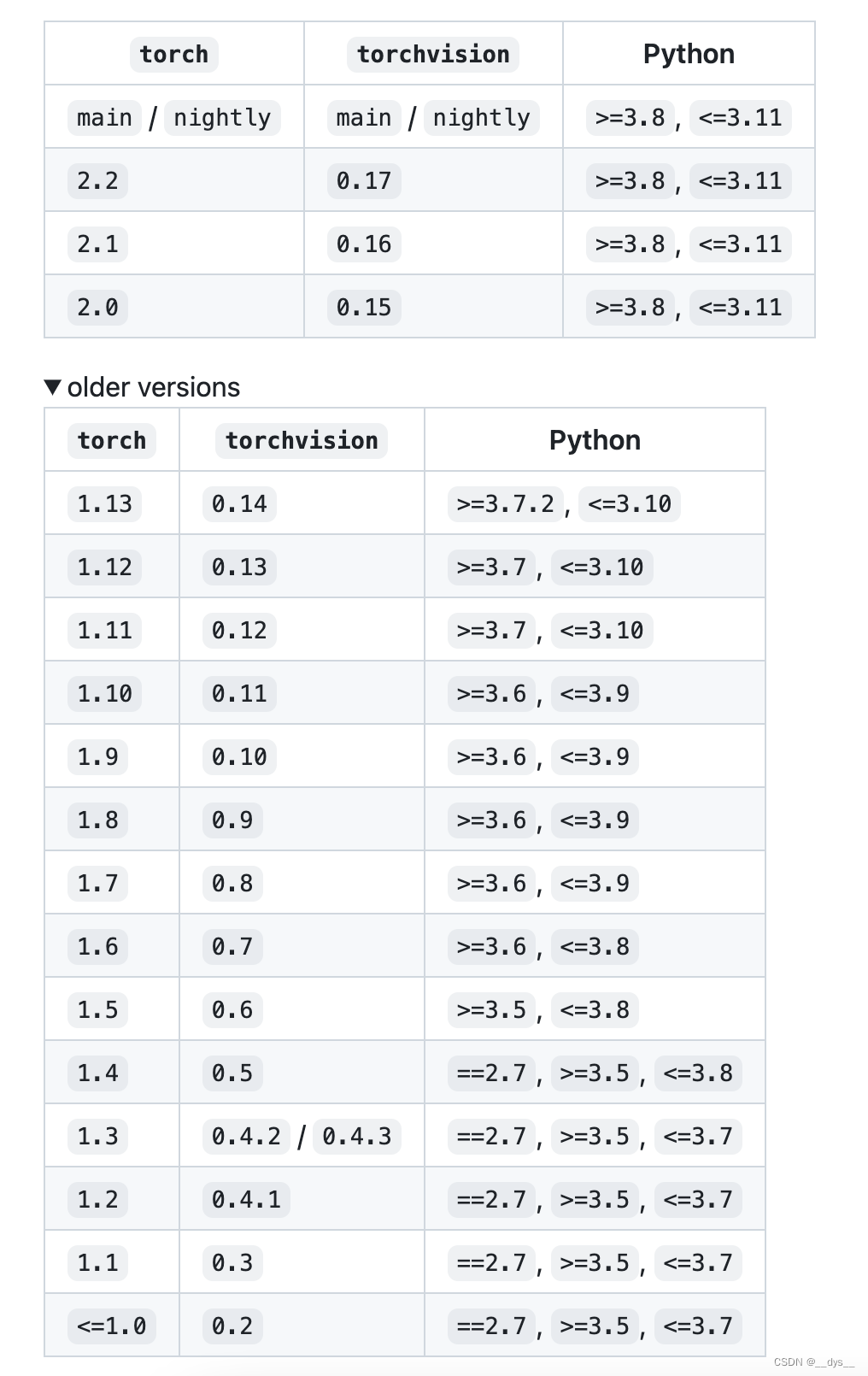
安装:选择python 3.8,CUDA 11.3,
git clone https://github.com/STOmics/STCellbin.git
mamba create --name=STCellbin python=3.8
conda activate STCellbin
# mamba install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 -c pytorch
mamba install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 cudatoolkit=11.3 -c pytorch
cd STCellbin
pip install -r requirements.txt -i https://pypi.tuna.tsinghua.edu.cn/simple
另外,还需要单独安装pyvips:
mamba install --channel conda-forge pyvips==2.2.1
运行
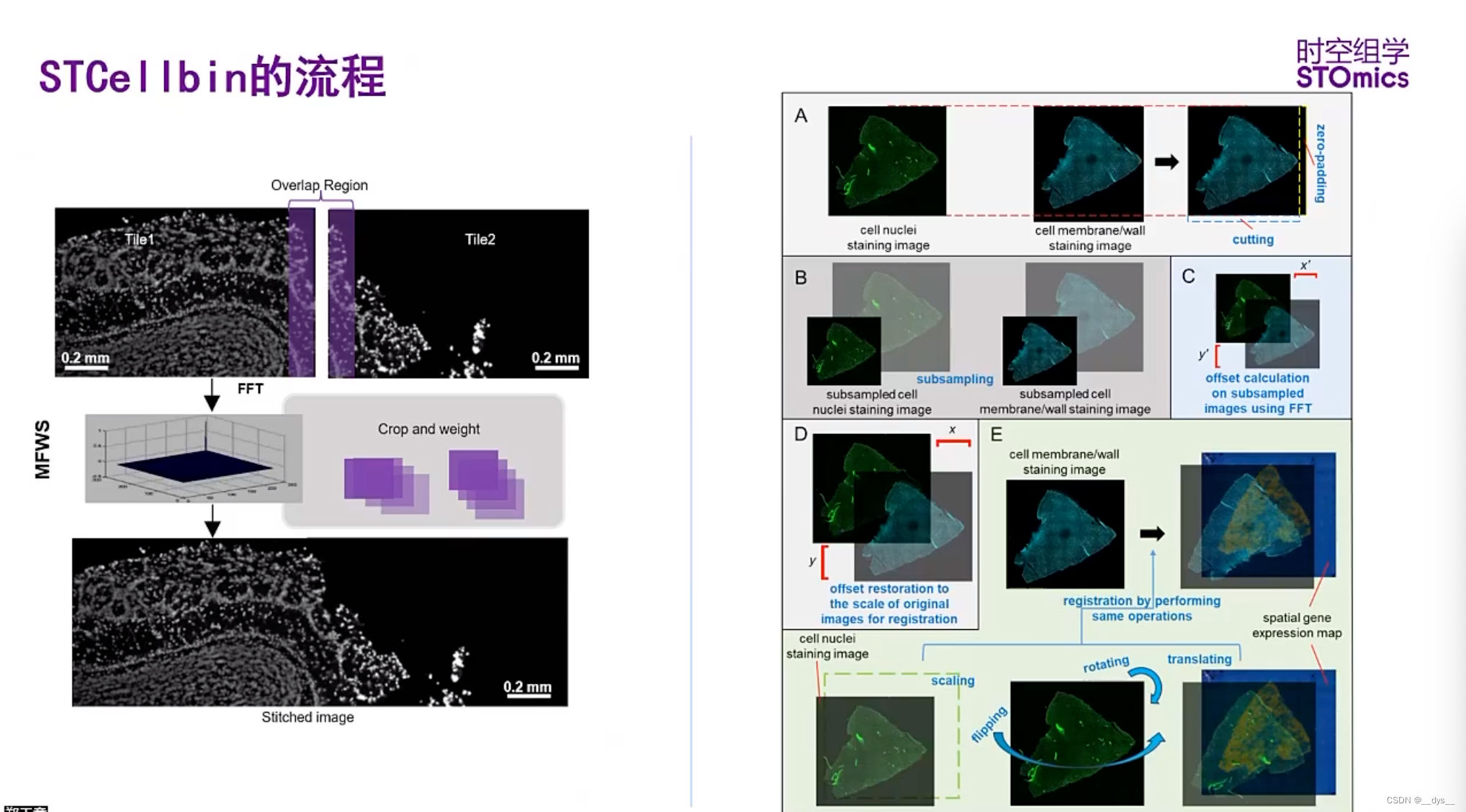
图像拼接和配准
- 显微镜扫描后形成多种小图,使用ImageStudio进行图像拼接。
- 图像配准:ssDNA图与gem配准;ssDNA图与细胞膜或者细胞壁荧光图配准。
下载测试数据
- 从CNGBdb数据库中,检索项目号STT0000048下载
- 开发者提供华大网盘:https://bgipan.genomics.cn/#/link/PNF9wOur6xawSfQYYdg2 密码:JlI9
有两个样本的数据C01344C4(小鼠肝脏,细胞膜)和FP200000449TL_C3(拟南芥种子,细胞壁),包括表达矩阵"gem.gz"和图片"image.tar"。下载完后,解压即可得到:
细胞核染色图,ssDNA
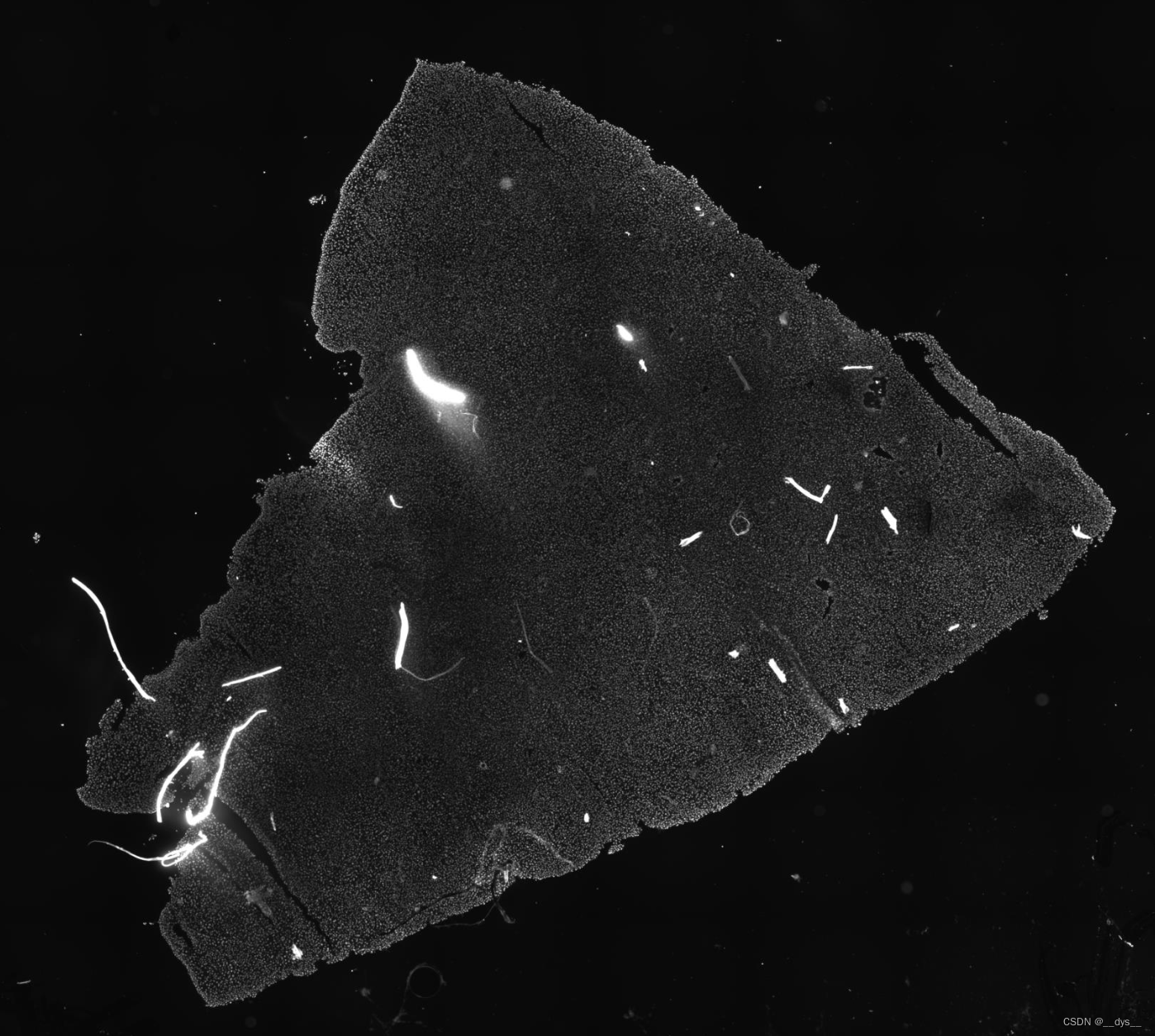
总体

局部
细胞膜颜色图(局部),DAPI
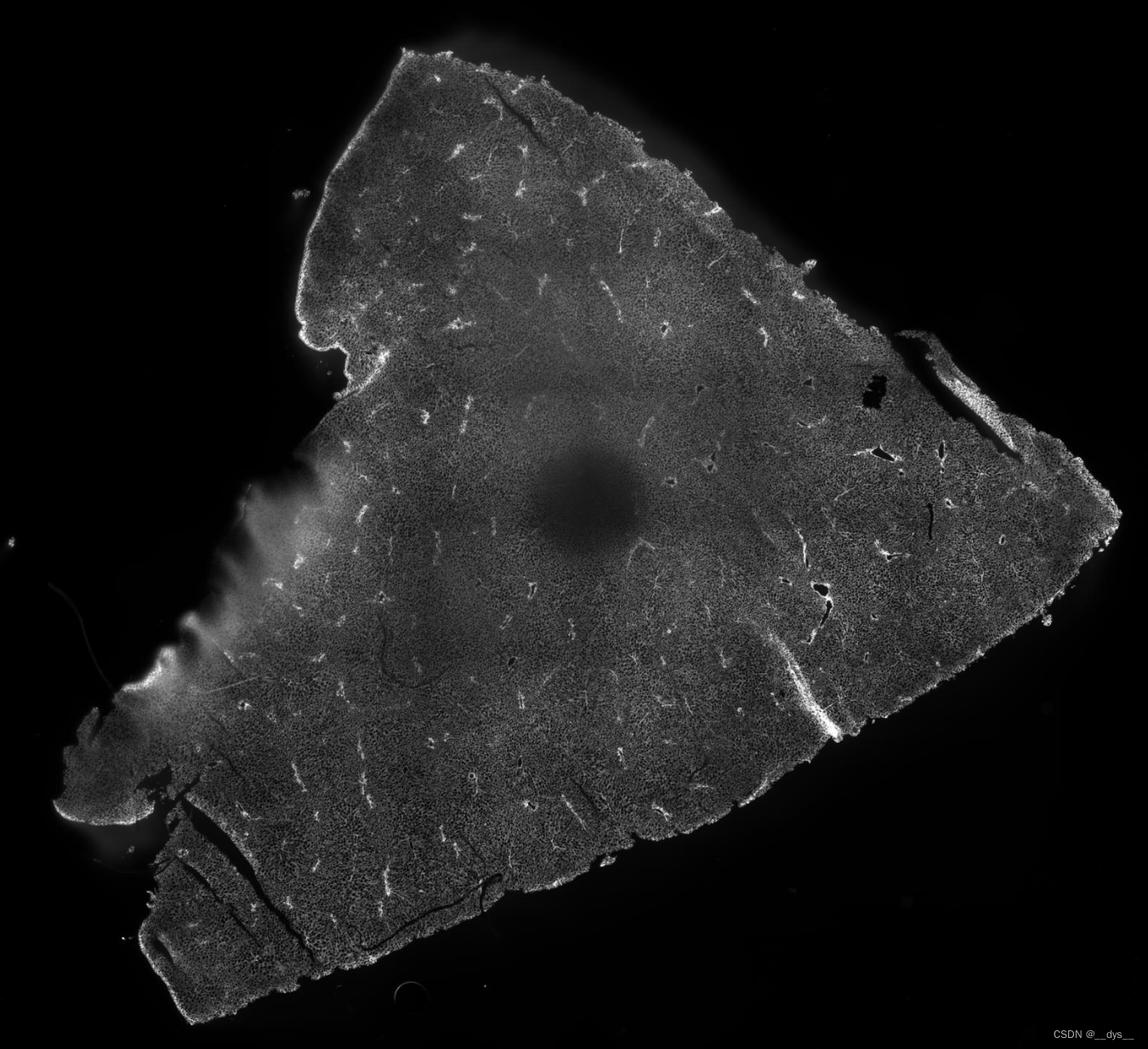
总体
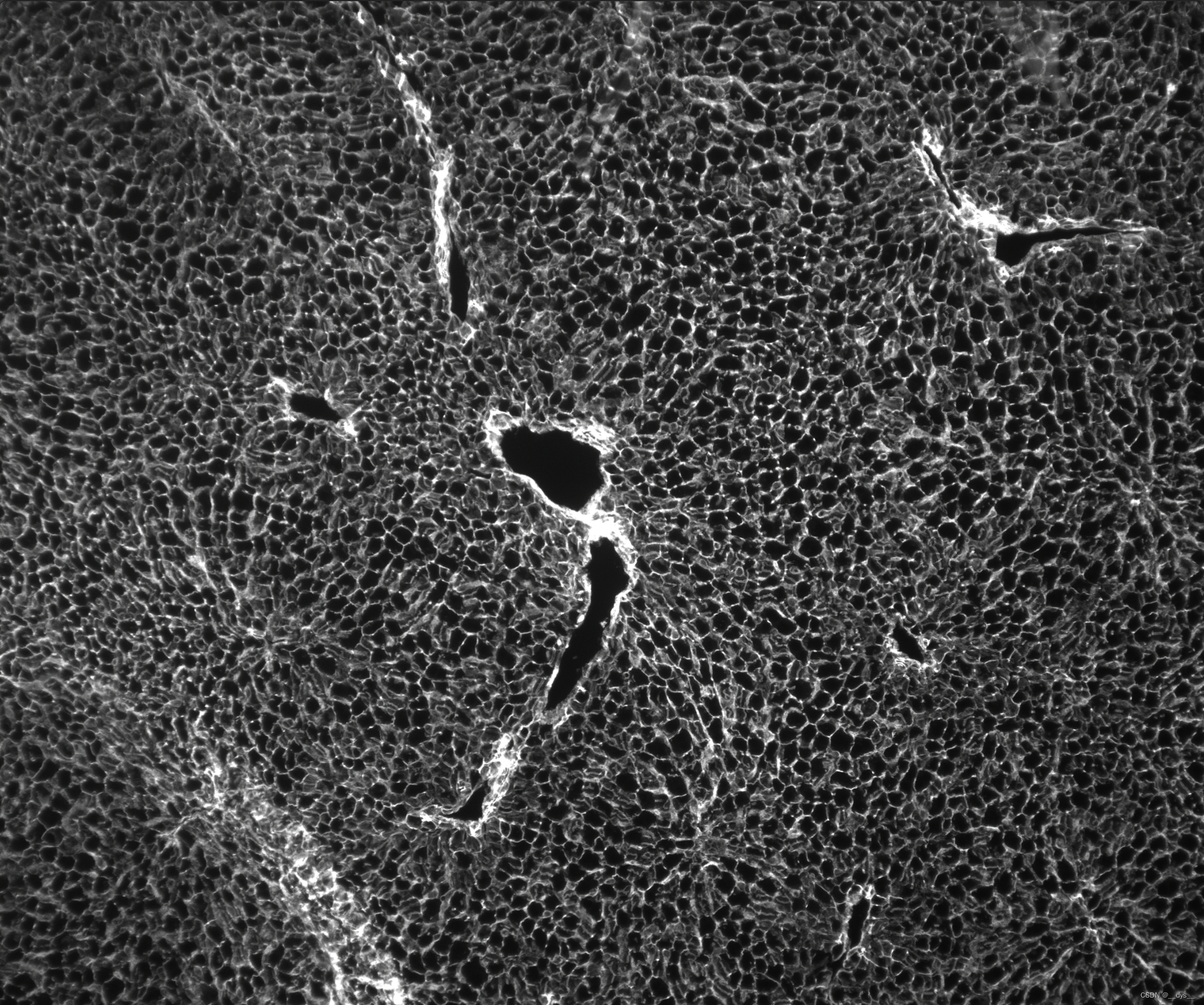
局部
运行
程序自动检测GPU,检测到则用GPU运行。则用GPU运行。由于demo数据是已经配准好的,所以可以直接运行。
python STCellbin/STCellbin.py \
-i /data/C01344C4,/data/C01344C4_Actin_IF \
-m /data/C01344C4.gem.gz \
-o /result \
-c C01344C4
可能遇到的问题
报错1:IndexError: list index out of range 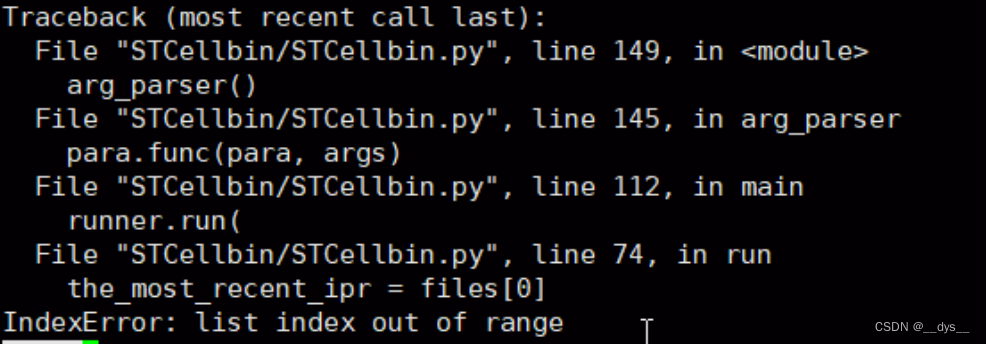
原因:可能由于文件识别问题
解决:将运行命令中的相对路径,改为绝对路径
python /home/dengysh/Project/STCellbin/STCellbin/STCellbin.py \
-i /home/dengysh/Project/STCellbin/data/C01344C4,/home/dengysh/Project/STCellbin/data/C01344C4_Actin_IF \
-m /home/dengysh/Project/STCellbin/data/C01344C4.gem.gz \
-o /home/dengysh/Project/STCellbin/result \
-c C01344C4
报错2:PackageNotFoundError: cell-bin 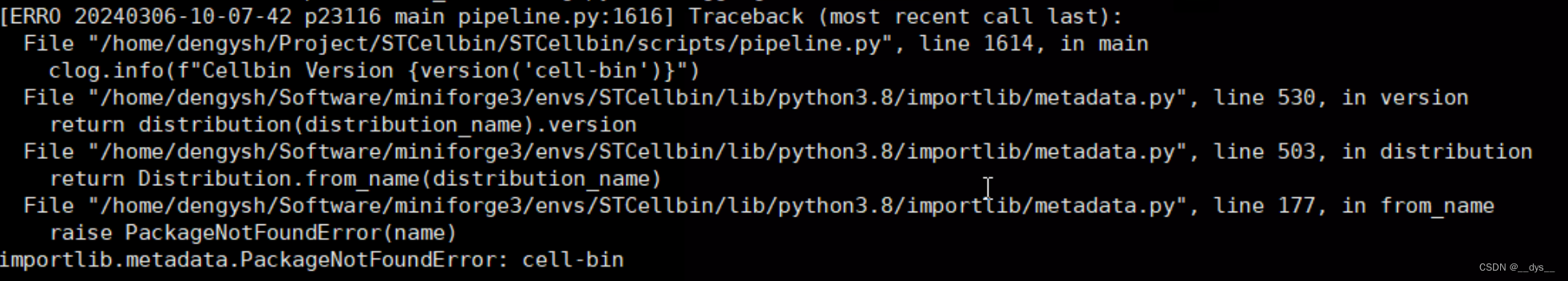
原因:缺少cell-bin模块。
解决:缺啥补啥,把cell-bin模块安装。pip install cell-bin -i https://pypi.tuna.tsinghua.edu.cn/simple
报错3:CUDA error
RuntimeError: CUDA error: CUBLAS_STATUS_INVALID_VALUE when calling `cublasSgemm( handle, opa, opb, m, n, k, &alpha, a, lda, b, ldb, &beta, c, ldc)`
原因:可能是共享库路径错误,
解决:unset LD_LIBRARY_PATH,不指定共性库路径,系统使用默认共享库路径
运行过程中出现警告信息:WARNING: no mask pixels found。但是最终结果也能正常输出。
CPU和GPU运行时间比较
GPU运行时间比CPU快90%
CPU大概运行了10个小时,GPU大概运行了1个小时。
## CPU
## 开始
[INFO 20240306-11-27-20 p29038 arg_parser STCellbin.py:129] 1.0.0
## 结束
[INFO 20240306-21-34-04 p37421 run_pipeline pipeline.py:1604] --------------------------------end------
C01344C4_regist_Actin
using device cpu
252372
uint8## GPU
## 开始
[INFO 20240308-21-46-34 p31943 arg_parser STCellbin.py:129] 1.0.0
## 结束
[INFO 20240308-22-51-49 p3653 run_pipeline pipeline.py:1604] --------------------------------end-------
C01344C4_regist_Actin
using device cuda:0
223639
uint8
输出
C01344C4
细胞分割前的输出文件: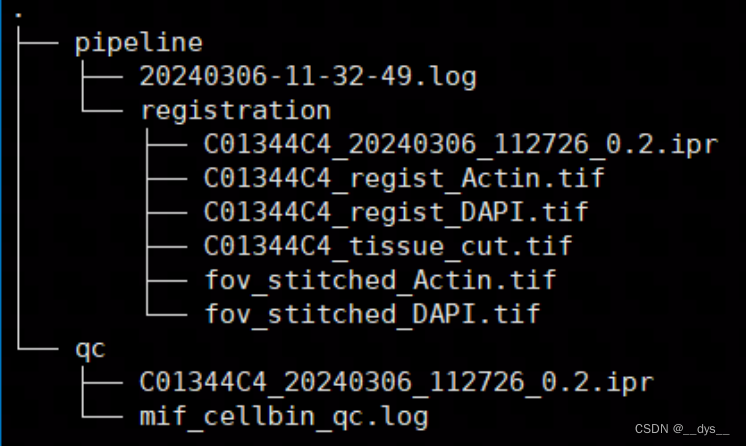
运行完成后,最终输出: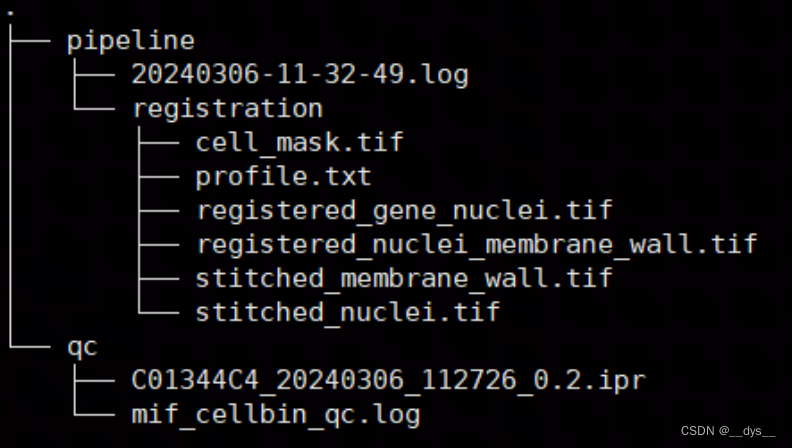
华大培训视频中显示分析过程中输出文件(比较模糊,主要是想对比输出是否一致):
流程分析完成后会进行文件删除: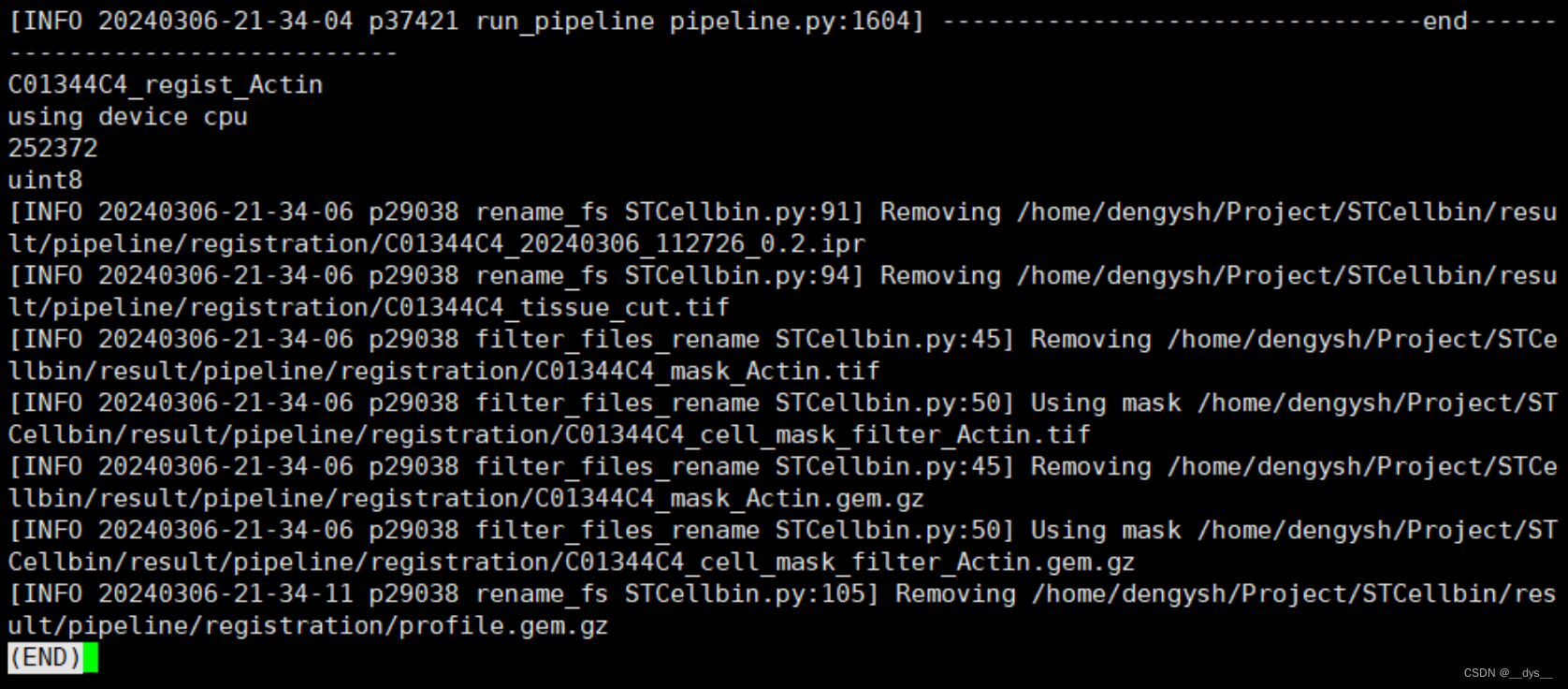
cell_mask.tif:细胞分割图像。Demo数据中间部分出现较大空缺,可能是由于细胞膜染色图模糊导致。 说明STCellBin对图像的质量要求还是挺高的,图像质量差的区域可能会无法进行细胞分割。
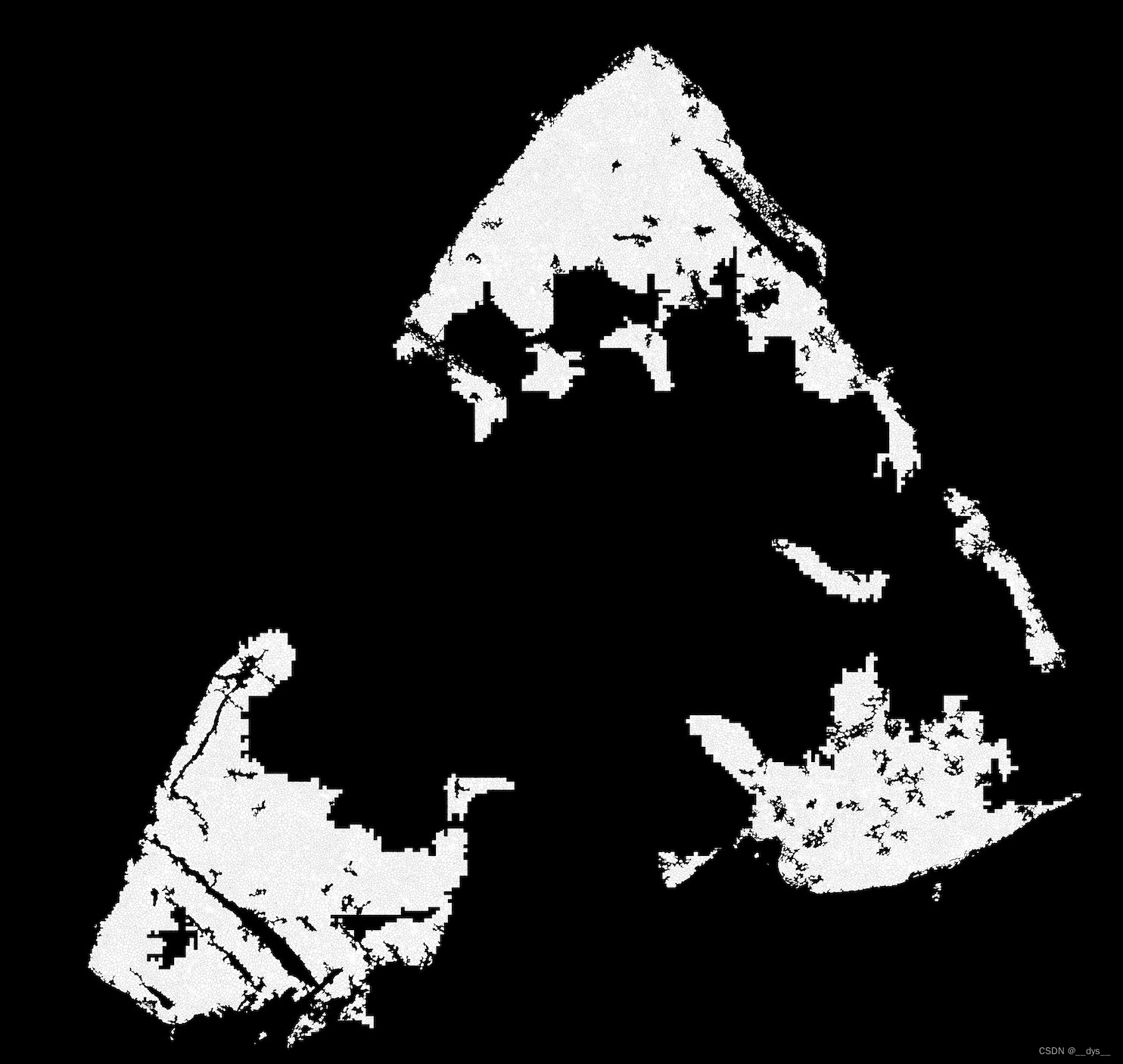
总体

局部
profile.txt:基因表达图谱gem。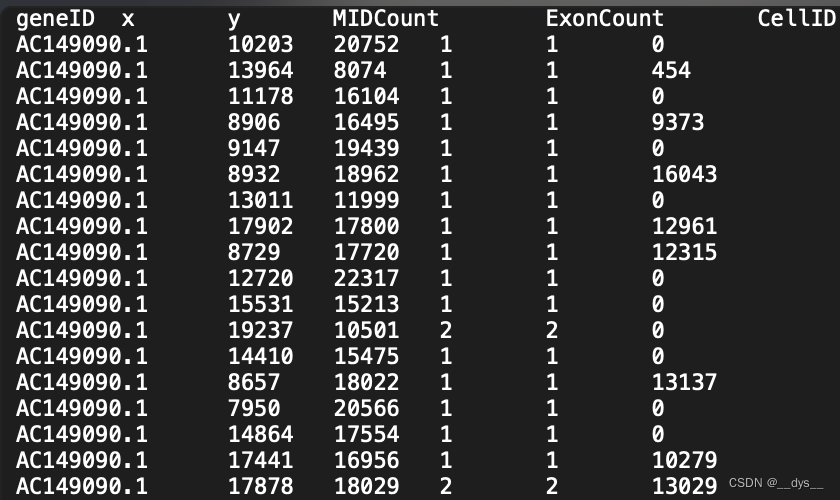
格式转换:GEM转scanpy/Seurat H5/RDS
GEM转成表达矩阵和细胞定位信息,再导出对应的H5文件
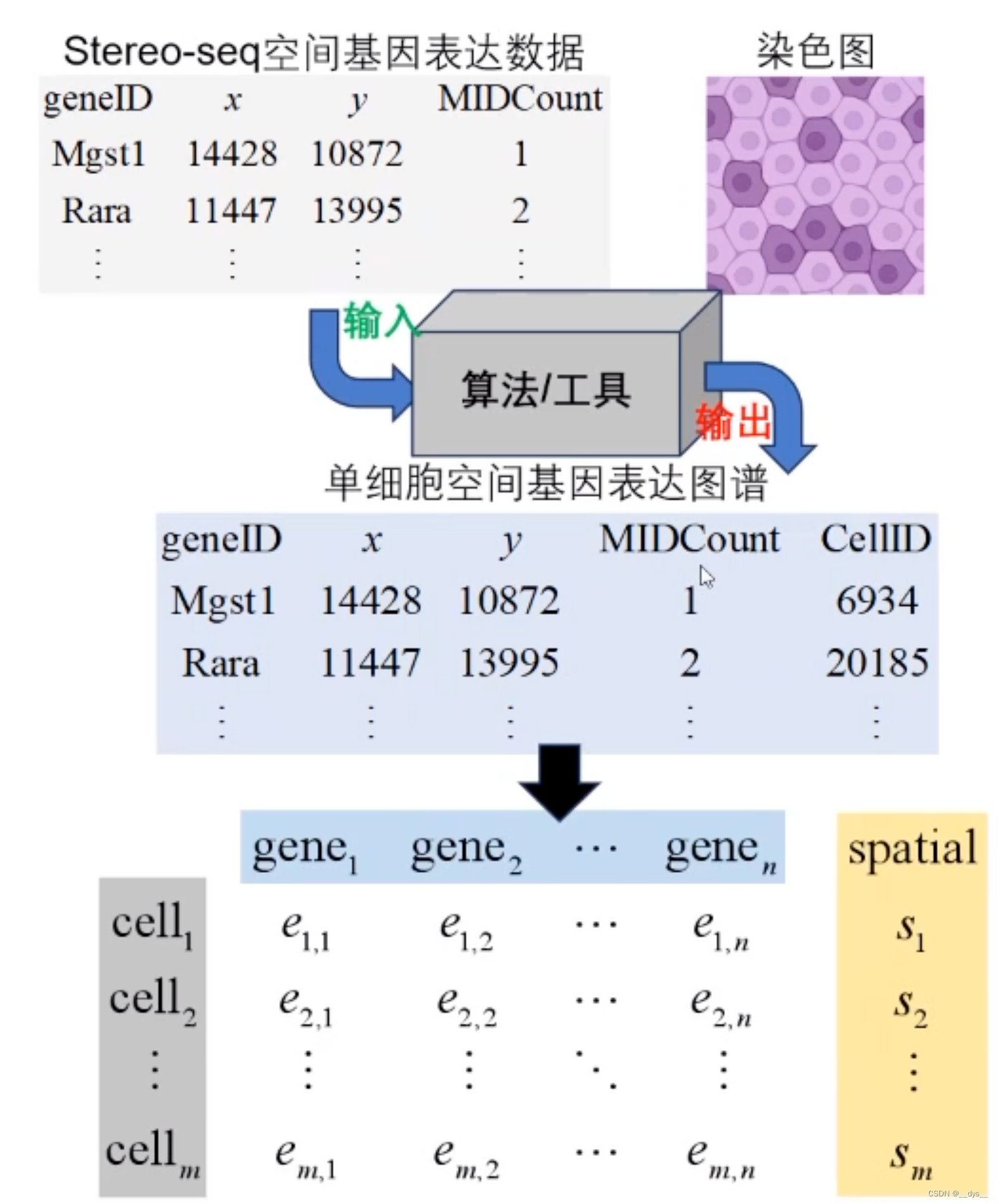
## 没安装Stereopy的,需要先安装
## mamba install stereopy -c stereopy -c grst -c numba -c conda-forge -c bioconda -c fastai
import stereo as st
import warnings
warnings.filterwarnings('ignore')# 读取Profile.txt,文件属于GEM文件,所以用read_gem导入
data_path = './result_GPU_C01344C4/pipeline/registration/profile.txt'
data = st.io.read_gem(file_path=data_path,sep='\t',bin_type='cell_bins',is_sparse=True,)## 查看data
data
## StereoExpData object with n_cells X n_genes = 17984 X 19863
## bin_type: cell_bins
## offset_x = 0
## offset_y = 0
## cells: ['cell_name']
## genes: ['gene_name']
## result: []## 做一些qc统计
data.tl.cal_qc()
data.tl.raw_checkpoint()
## data
## StereoExpData object with n_cells X n_genes = 17984 X 19863
## bin_type: cell_bins
## offset_x = 0
## offset_y = 0
## cells: ['cell_name', 'total_counts', 'n_genes_by_counts', 'pct_counts_mt']
## genes: ['gene_name', 'n_cells', 'n_counts', 'mean_umi']
## result: []## 导出h5文件,可用scanpy做后续分析
adata = st.io.stereo_to_anndata(data,flavor='scanpy',output='scanpy_out.h5ad')## 也可以导出Seurat的h5
adata = st.io.stereo_to_anndata(data,flavor='seurat',output='seurat_out.h5ad')## H5ad转RDS
### 新脚本 h5ad2rds.R
Rscript h5ad2rds.R --infile seurat_out.h5ad --outfile seurat_out.rds
### 旧脚本 annh5ad2rds.R
Rscript annh5ad2rds.R --infile seurat_out.h5ad --outfile seurat_out.rds
注意:Stereopy提供了转换脚本h5ad2rds.R,该脚本更新后,注释了image的写入,所以rds中无image信息,运行部分函数可能会报错(SpatialFeaturePlot等)。

FP200000449TL_C3

局部
StereoCell:基于细胞核分割
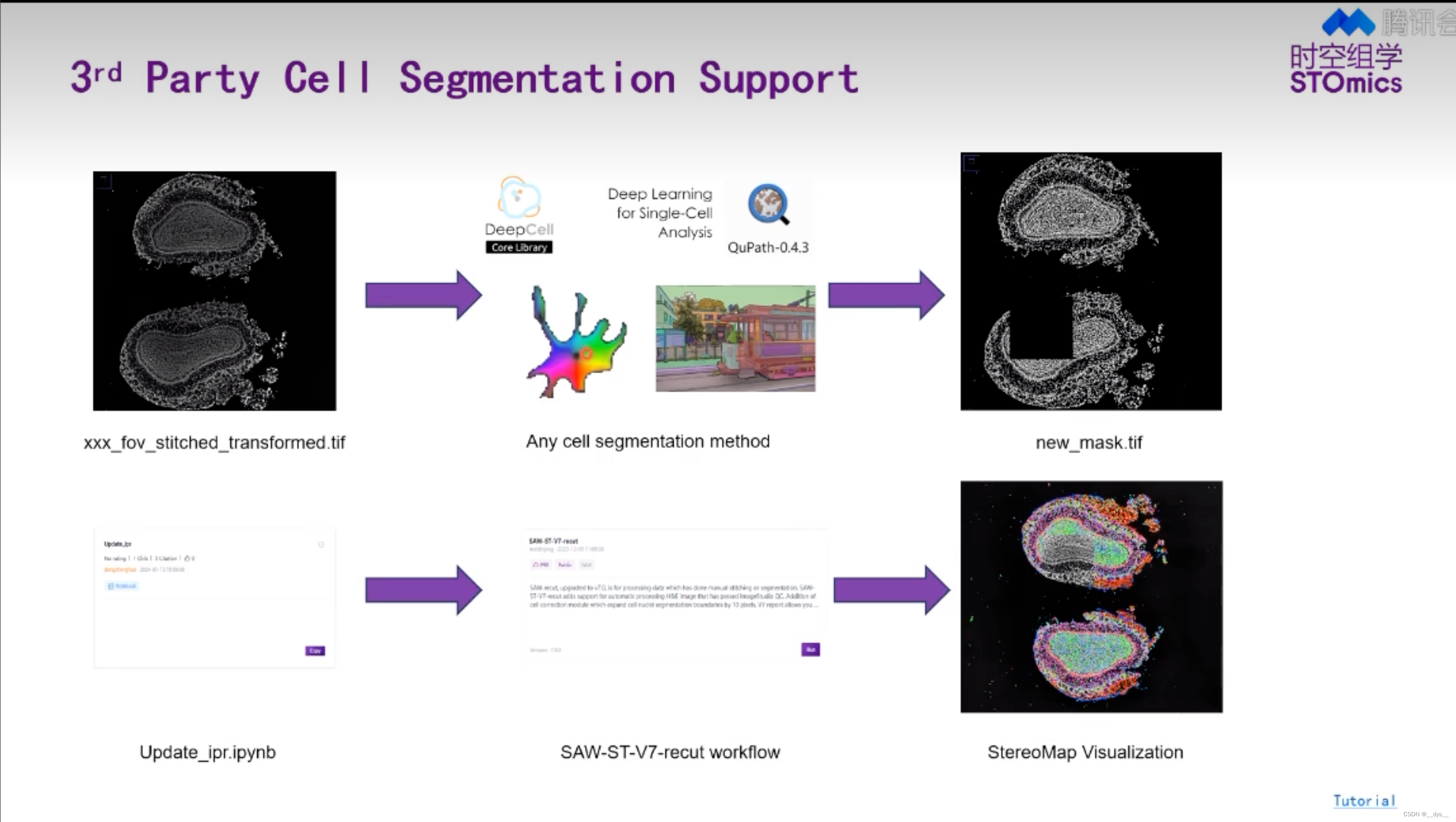
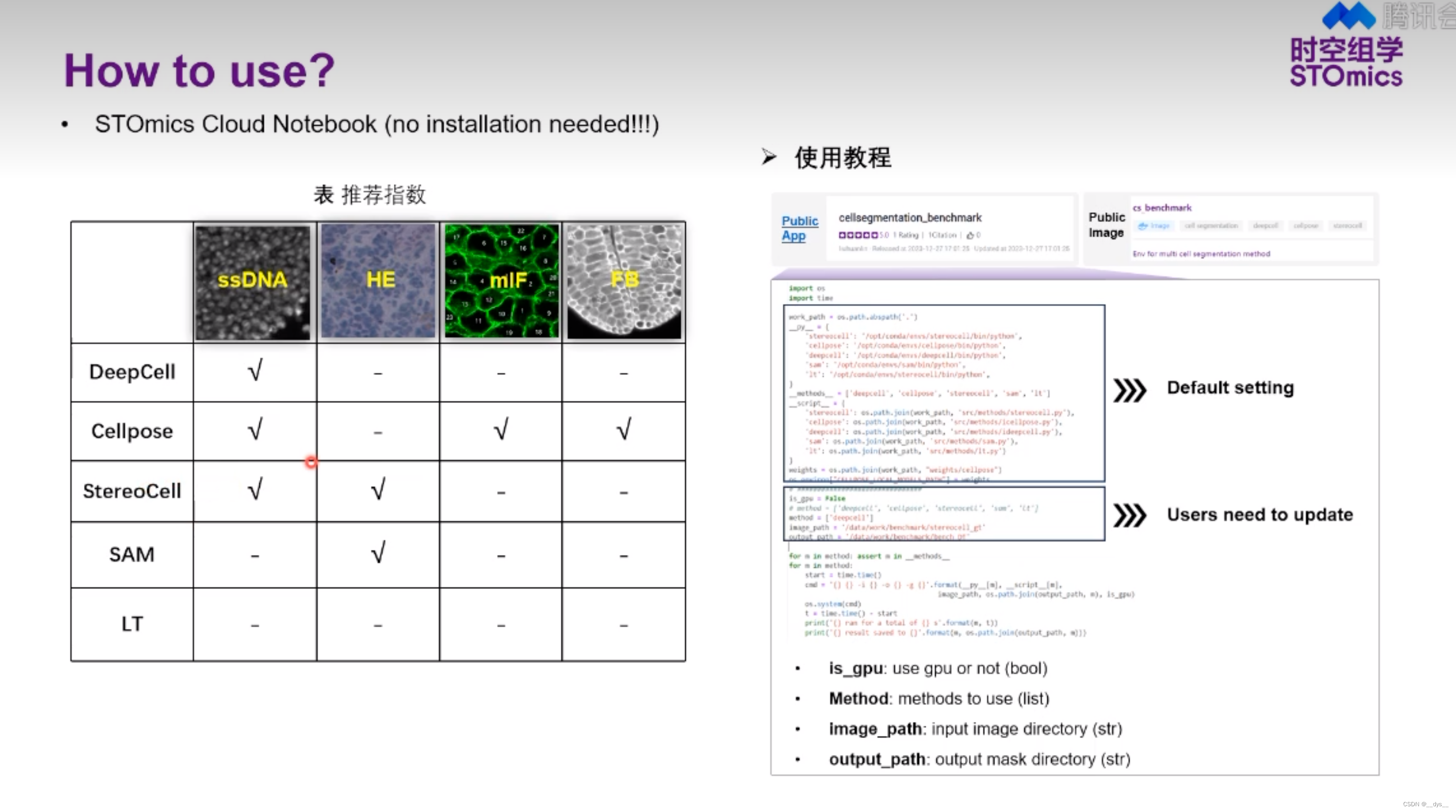
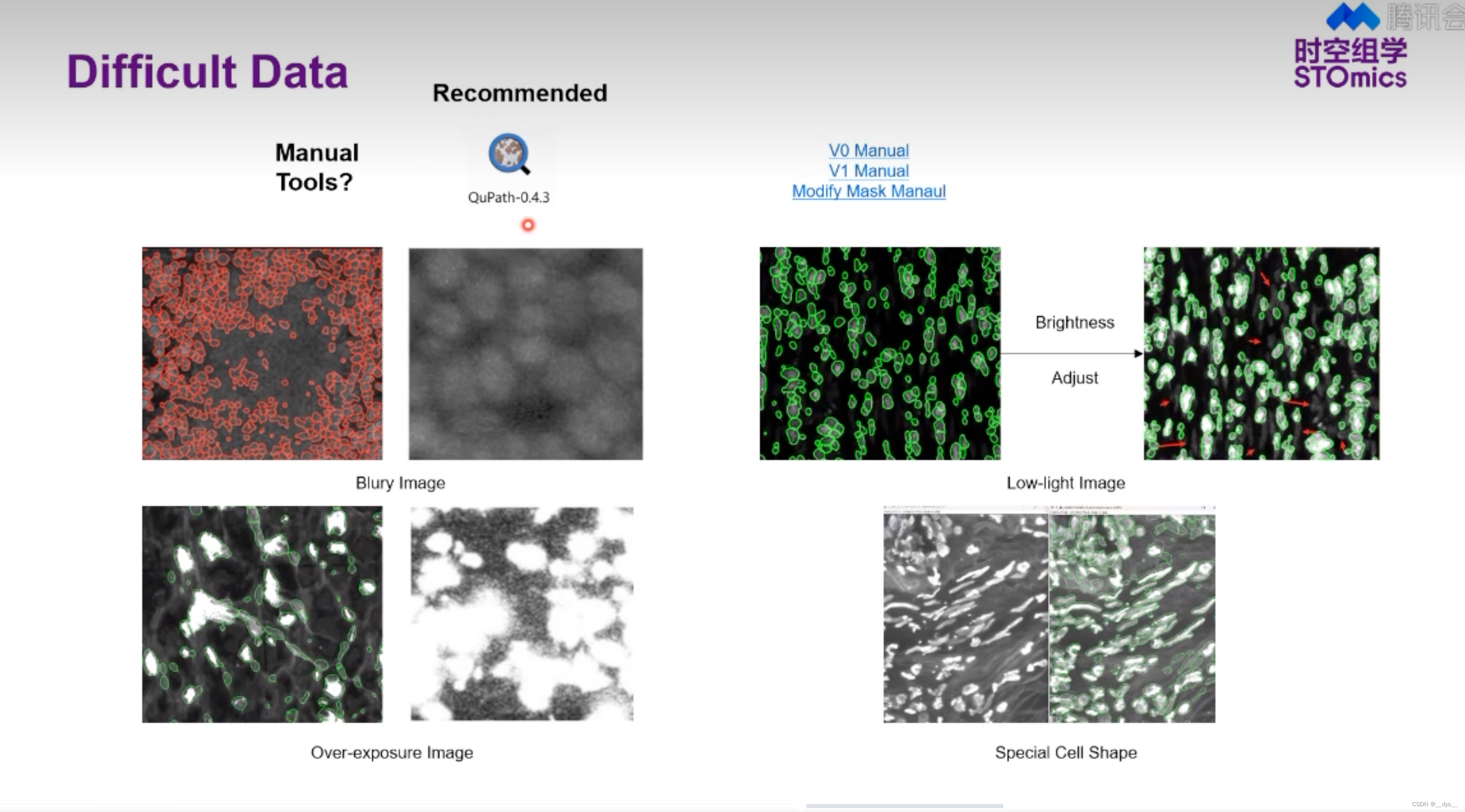
- SAW流程可进行Cellbin,获得基于细胞核预测的细胞分割结果Cellbin。
- 可以使用第三方工具进行细胞分割,获得mask后,再使用SAW进行分析,得到细胞分割结果。
补充材料
H5ad转RDS,旧脚本annh5ad2rds.R
library(dplyr)
library(rjson)
library(Seurat)
library(ggplot2)
library(argparser)
library(SeuratDisk)args <- arg_parser("Converting h5ad file(.h5ad) to RDS.")
args <- add_argument(args, "--infile", help = "input .h5ad file")
args <- add_argument(args, "--outfile", help = "output RDS file")
argv <- parse_args(args)if ( is.null(argv$infile) || is.null(argv$outfile) ) {print('positional argument `infile` or `outfile` is null')quit('no', -1)
}infile <- argv$infile
outfile <- argv$outfile# convert h5ad as h5seurat, which means a seurat-object format stored in h5
Convert(infile, dest = "h5seurat", assay = "Spatial", overwrite = TRUE)h5file <- paste(paste(unlist(strsplit(infile, "h5ad", fixed = TRUE)), collapse='h5ad'), "h5seurat", sep="")
print(paste(c("Finished! Converting h5ad to h5seurat file at:", h5file), sep=" ", collapse=NULL))object <- LoadH5Seurat(h5file)
print(paste(c("Successfully load h5seurat:", h5file), sep=" ", collapse=NULL))# spatial already transform to `Spatial`` in assays
if (!is.null(object@reductions$spatial)) {object@reductions$spatial <- NULL
}# convert stereopy SCT result to seurat SCT result
if (!is.null(object@misc$sct_counts) &&!is.null(object@misc$sct_data) &&!is.null(object@misc$sct_scale) &&!is.null(object@misc$sct_cellname) &&!is.null(object@misc$sct_genename) &&!is.null(object@misc$sct_top_features)) {sct.assay.out <- CreateAssayObject(counts=object[['Spatial']]@counts, check.matrix=FALSE)# VariableFeatures(object=sct.assay.out) <- rownames(object@misc$sct_top_features)sct.assay.out <- SetAssayData(object = sct.assay.out,slot = "data",new.data = log1p(x=GetAssayData(object=sct.assay.out, slot="counts")))sct.assay.out@scale.data <- as.matrix(object@misc$sct_scale)colnames(sct.assay.out@scale.data) <- object@misc$sct_cellnamerownames(sct.assay.out@scale.data) <- object@misc$sct_top_featuressct.assay.out <- Seurat:::SCTAssay(sct.assay.out, assay.orig='Spatial')Seurat::VariableFeatures(object = sct.assay.out) <- object@misc$sct_top_featuresobject[['SCT']] <- sct.assay.outDefaultAssay(object=object) <- 'SCT'# TODO: tag the reductions as SCT, this will influence the find_cluster choice of dataobject@reductions$pca@assay.used <- 'SCT'object@reductions$umap@assay.used <- 'SCT'assay.used <- 'SCT'print("Finished! Got SCTransform result in object, create a new SCTAssay and set it as default assay.")
} else {# TODO: we now only save raw counts, try not to add raw counts to .data, do NormalizeData to fit thisobject <- NormalizeData(object)assay.used <- 'Spatial'print("Finished! Got raw counts only, auto create log-normalize data.")
}# TODO follow with old code, don't touch
print("Start add image...This may take some minutes...(~.~)")
# add image
cell_coords <- unique(object@meta.data[, c('x', 'y')])
cell_coords['cell'] <- row.names(cell_coords)
cell_coords$x <- cell_coords$x - min(cell_coords$x) + 1
cell_coords$y <- cell_coords$y - min(cell_coords$y) + 1# object of images$slice1@image, all illustrated as 1 since no concrete pic
tissue_lowres_image <- matrix(1, max(cell_coords$y), max(cell_coords$x))# object of images$slice1@coordinates, concrete coordinate of X and Y
tissue_positions_list <- data.frame(row.names = cell_coords$cell,tissue = 1,row = cell_coords$y, col = cell_coords$x,imagerow = cell_coords$y, imagecol = cell_coords$x)
# @images$slice1@scale.factors
scalefactors_json <- toJSON(list(fiducial_diameter_fullres = 1, tissue_hires_scalef = 1, tissue_lowres_scalef = 1))# generate object @images$slice1
generate_BGI_spatial <- function(image, scale.factors, tissue.positions, filter.matrix = TRUE) {if (filter.matrix) {tissue.positions <- tissue.positions[which(tissue.positions$tissue == 1), , drop = FALSE]}unnormalized.radius <- scale.factors$fiducial_diameter_fullres * scale.factors$tissue_lowres_scalefspot.radius <- unnormalized.radius / max(dim(x = image))return(new(Class = 'VisiumV1',image = image,scale.factors = scalefactors(spot = scale.factors$tissue_hires_scalef,fiducial = scale.factors$fiducial_diameter_fullres,hires = scale.factors$tissue_hires_scalef,lowres = scale.factors$tissue_lowres_scalef),coordinates = tissue.positions,spot.radius = spot.radius))
}BGI_spatial <- generate_BGI_spatial(image = tissue_lowres_image,scale.factors = fromJSON(scalefactors_json),tissue.positions = tissue_positions_list)# can be thought of as a background of spatial
# import image into seurat object
object@images[['slice1']] <- BGI_spatial
object@images$slice1@key <- "slice1_"
object@images$slice1@assay <- assay.used# do not use these code if you know what you wanna do
# ---log-normalize
#object <- FindVariableFeatures(object, selection.method = "vst", nfeatures = 2000)
#all.genes <- rownames(object)
#object <- ScaleData(object, features = all.genes)
# ---log-normalize rest part / sctransform
#object <- RunPCA(object, verbose = FALSE)
#object <- RunUMAP(object, dims = 1:20, verbose = FALSE)
#object <- FindNeighbors(object, dims = 1:20, verbose = FALSE)
#object <- FindClusters(object, verbose = FALSE)
#object <- DimPlot(object, label = FALSE) + NoLegend()
#print('Test finished')# conversion done, save
print("Finished add image...Start to saveRDS...")
saveRDS(object, outfile)
print("Finished RDS.")
quit('yes', 0)
参考资料
- Zhang B, Li M, Kang Q, et al. Generating single-cell gene expression profiles for high-resolution spatial transcriptomics based on cell boundary images. GigaByte. 2024;2024:gigabyte110.
- STOmics/STCellbin: Enhanced application on generating single-cell gene expression profile for high-resolution spatial transcriptomics. (github.com)
- python pytorch-GPU 环境搭建 (CUDA 11.2)_cuda11.2对应的pytorch-CSDN博客
- 华大时空,视频号,培训视频:解码数据:前沿算法工具赋能时空组学研究