1.bwa的下载与安装
https://www.jianshu.com/p/19f58a07e6f4
主要参考这篇帖子,如果之前的步骤都走通了的话,依赖什么的不用特别安装,报错了再补也可以
安好了之后,进到他的路径,输./bwa,就可以确认bwa有没有安装好了,环境设置好以后可以在其他地方输入bwa,也可以叫他出来。
在正式开始使用之前,注意建立索引index:
Usage:bwa index [ –p prefix ] [ –a algoType ] <in.db.fasta>
示例:
解释:
–p prefix :这是你的index的名字,或者说前缀,这个名字是你自己起的,我叫他ZS97genome
–a algoType :-a [is|bwtsw] 构建index的算法,有以下两个选项:
-a is 是默认的算法,虽然相对较快,但是需要较大的内存,当构建的数据库大于2GB的时候就不能正常工作了;
-a bwtsw 对于短的参考序列式不工作的,必须要大于等于10MB, 但能用于较大的基因组数据,比如人的全基因组。
<in.db.fasta>:这个是你先下载的基因组文件,记得写绝对路径
建立的过程就是这样,显示这样的东西就说明建立好了:
2.利用bwa将数据与参考基因组进行比对,获得sam文件
这里主要是使用bwa的 BWA-MEM 算法,输入命令:
Usage: bwa mem [options] ref.fa reads.fq [mates.fq]
解释范例:
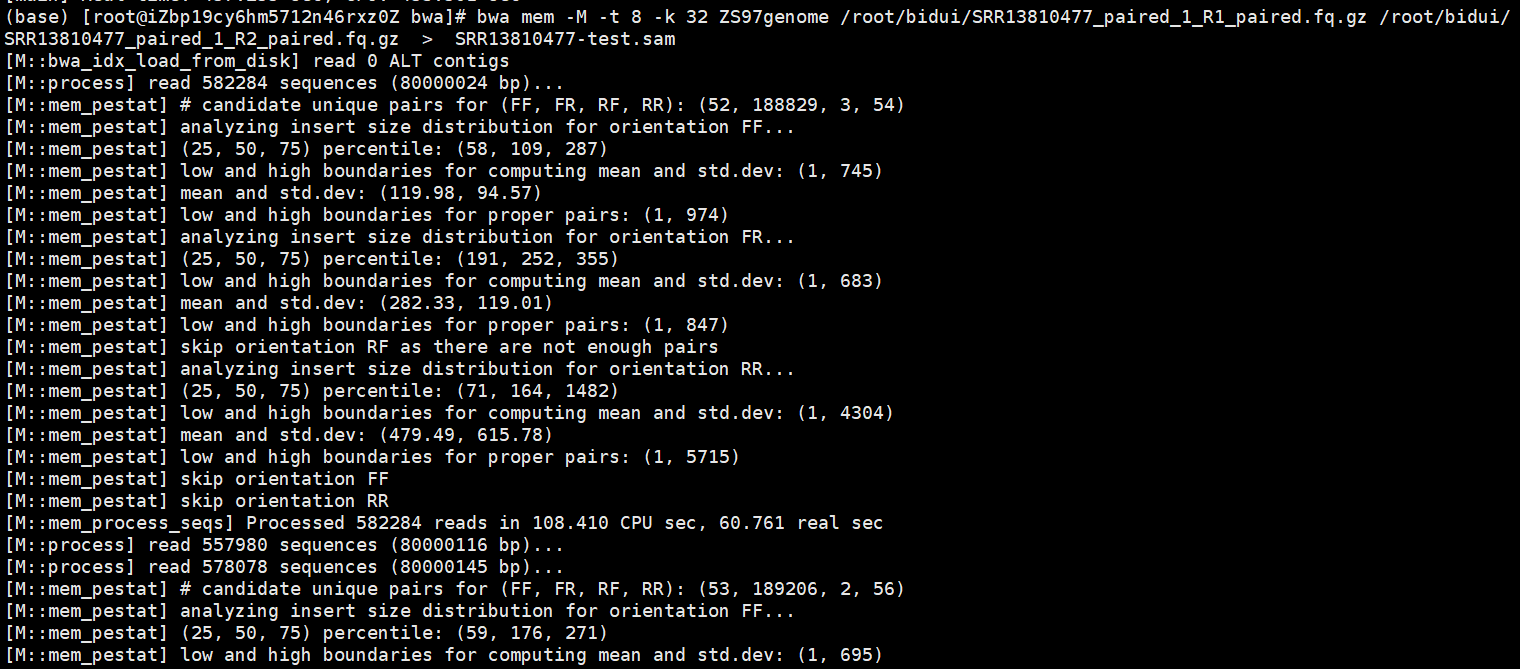
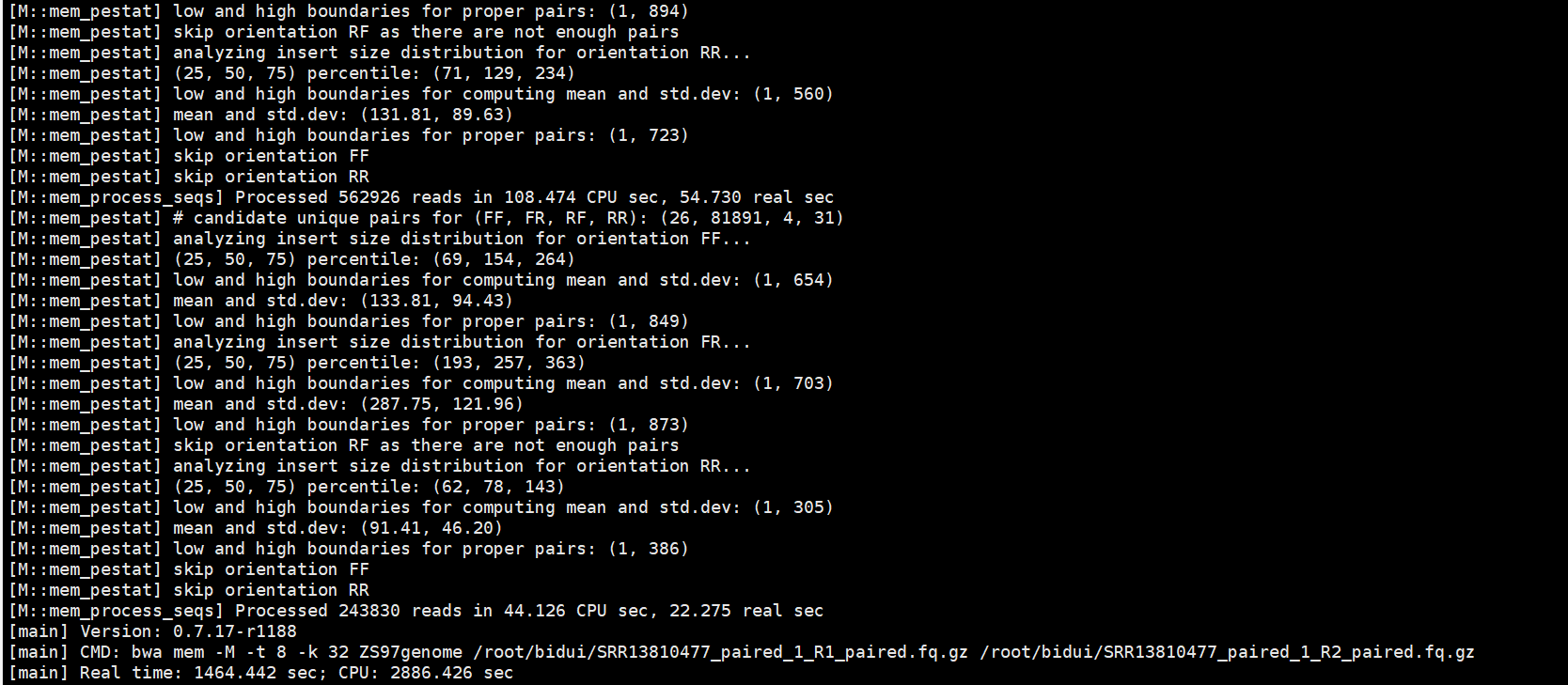
bwa mem -M -t 8 -k 32 ZS97genome /root/bidui/SRR13810477_paired_1_R1_paired.fq.gz /root/bidui/SRR13810477_paired_1_R2_paired.fq.gz > SRR13810477-test.sam
[options]:这是一些按需求调节的参数,主要如下:
-t INT 线程数,默认是1,增加线程数,会减少运行时间。
-M 将 shorter split hits 标记为次优,以兼容 Picard’s markDuplicates 软件。
-p 若无此参数:输入文件只有1个,则进行单端比对;若输入文件有2个,则作为paired reads进行比对。若加入此参数:则仅以第1个文件作为输入(会忽略第二个输入序列文件,把第一个文件当做单端测序的数据进行比对),该文件必须是read1.fq和read2.fa进行reads交叉的数据。
-R STR 完整的read group的头部,可以用 '\t' 作为分隔符, 在输出的SAM文件中被解释为制表符TAB. read group 的ID,会被添加到输出文件的每一个read的头部。
-T INT 当比对的分值比 INT 小时,不输出该比对结果,这个参数只影响输出的结果,不影响比对的过程。
-a 将所有的比对结果都输出,包括 single-end 和 unpaired paired-end的 reads,但是这些比对的结果会被标记为次优。
-Y 对数据进行soft clipping, 当错配或者gap数过多比对不上时,会对序列进行切除,这里的切除并只是在比对时去掉这部分序列,最终输出结果中序列还是存在的,所以称为soft clipping。
ref.fa:这是你刚刚建立的index的名字
reads.fq:这是你用Trimmomatic处理出来的数据,记得用绝对路径
SRR13810477-test.sam:这就是你要输出的文件的文件名
特别说明:
如果 mates.fq 缺省,且参数 –p 未设定,那么 reads.fq 被认为是 single-end;
如果 mates.fq 存在,且参数 –p 未设定,那么 mem 命令会认为 read.fq 和 mates.fq 中的 i-th reads 组成一个read对 (a read pair),这个模式是常用的 paired-end mode。
如果参数 –p 被设定,那么, mem 命令会认为 read.fq 中的 第 2i-th 和 第 (2i + 1)-th 的 reads 组成一个 read 对 (a read pair),这种方式也被成为交错式的(interleaved paired-end)。 在这种情况下,即使有 mates.fq,也会被忽略。
过程主要是这个样子的:
处理完就会产生你想要的sam文件,当然也可以和samtools联用直接变成bam,不过第一次尝试就一步一步来吧
你可以在bwa的文件夹里找到你的sam文件,找不到就用find来找:
find / -name SRR13810477-test.sam