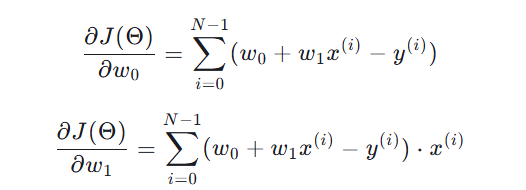用于药物相互作用预测的可解释多视图注意网络
论文标题: Interpretable multi-view attention network for drug-drug interaction prediction
论文地址: https://ieeexplore.ieee.org/document/10385757
论文期刊: BIBM 2024
摘要
药物间相互作用(DDI)在药物发现中发挥着越来越重要的作用。预测潜在的 DDI 对于临床研究也至关重要。考虑到湿实验室实验的高成本和风险,计算机 DDI 预测是一种替代选择。最近,深度学习方法已被开发用于 DDI 预测。然而,大多数现有方法侧重于从分子 SMILES 序列或药物交互网络中提取特征,忽略了可以从这两种视图中得出的有价值的补充信息。在本文中,我们提出了一种用于 DDI 预测的新型可解释多视图注意力网络(MVA-DDI)。 MVA-DDI可以有效地从不同角度提取药物表征,以改进DDI预测。具体来说,对于给定的药物,我们设计了一个基于变压器的编码器和一个基于图卷积网络的编码器,分别从 SMILES 序列和分子图学习序列和图表示。为了充分利用序列和分子视图之间的互补信息,进一步采用注意机制,通过考虑不同视图的重要性来自适应聚合序列和图形表示,生成最终的药物表示。比较实验表明,我们的 MVA-DDI1 模型在 DDI 预测方面取得了优于最先进模型的性能。
1.引言
药物相互作用(DDI)是指服用一种药物影响人体内另一种或多种药物的情况。这种相互作用可以是协同的,也可以产生全新的效果。由于单一药物能够治愈的疾病很少,因此多种药物联合治疗对于大多数疾病都比单一治疗更有效,这间接导致了具有副作用的DDI的出现。因此,DDI预测在药物发现和临床研究中至关重要。
传统上,DDI预测是通过广泛的生物学或药理学试验来进行的,这是耗时且费力的。随着大规模生物医学数据集的出现和人工智能技术的进步,深度学习模型已成为广泛的化学信息学和生物信息学领域的新兴范例。与此同时,深度学习方法在 DDI 预测中的应用激增,它可以作为一种低成本但有效的替代方案,通过从各种数据格式中提取药物特征来预测潜在的 DDI。以前的方法通常集中于获取药物 SMILES 和其他属性概况之间的相似特征。随着图神经网络(GNN)的广泛成功,近年来出现了提取分子图或子结构的拓扑特征的趋势。沿着这一趋势,研究人员提出基于 CASTER 等药物的化学结构数据进行 DDI 预测。另一方面,在KGNN中,融合图卷积网络(GCN)的思想来提取知识图谱的高阶结构和语义关系。
在大多数现有方法中,研究人员提出了一个单一网络来从单一角度学习分子表示,无论是基于相似性还是基于图(网络)的方法。然而,由于不同类型的 DDI 任务下的单视图学习,这些方法会遇到特征编码不足的问题。挑战在于,多个视角可能会相互干扰,特别是当从同一药物的不同视角学到的特征可以相互补充时。为了缓解这一挑战,我们提出了一种用于 DDI 预测的可解释多视图注意力网络(MVA-DDI)。
在所提出的模型中,我们使用 SMILES 序列作为单一输入,同时考虑来自同一药物分子的序列和分子图。具体来说,我们首先充分考虑药物 SMILES 之间的子结构,采用具有可解释子结构分区指纹(ESPF)编码的序列编码器(Transformer)。其次,我们使用图编码器(GCN)通过 RDKit 学习从 SMILES 转换而来的分子图来提取结构特征。同时,我们利用自我监督学习,通过设计分子图正负样本之间的对比损失函数来获得更多信息丰富的初始药物特征。之后,我们整合每种药物的序列和图特征,使用可解释的自注意力机制获得最终特征,该机制用于通过解码器预测药物对的潜在相互作用概率。我们在真实数据集上评估了我们提出的模型,实验结果表明我们的 MVA-DDI 优于基线方法。
本文的主要贡献总结如下:
- 我们提出了一种新颖的多视图注意力模型MVA-DDI来预测DDI,该模型有利于通过考虑SMILES序列和分子结构图来学习高质量的特征。
- MVA-DDI 设计了一个可解释的注意力网络,自适应地结合从不同视图中学习到的表示,这有助于提高 DDI 的预测性能。
- 真实世界数据集的实验结果证明了 MVA-DDI 相对于最先进方法的优越性。
2.相关工作
2.1 单视图学习
基于图嵌入的方法 复杂的数据结构,例如异构网络,是一个天然的高维空间,从这种复杂的结构中挖掘有效信息被认为是提高模型性能的替代方法。为了应对这一挑战,人们提出了基于图嵌入的方法,采用流行的网络嵌入算法来捕获网络的底层结构并导出潜在有效的基于网络的特征。这些方法可以大致分为基于矩阵分解的方法、基于随机游走的方法和基于神经网络的方法。然而,这方面的工作只关注节点之间的连接,而忽略了节点属性和边的类型。
基于知识图的方法 为了克服基于图嵌入的方法带来的知识缺乏,基于知识图(KG)的方法因其强大的异构数据表达能力而受到越来越多的关注。KG-DDI是第一个专门用于DDI预测的模型,它使用各种嵌入方法将节点嵌入到构建的 KG 中。近年来,随着图神经网络的流行,一些新颖的知识图谱方法融合了图卷积网络(GCN)的思想来提取知识图谱的高阶结构和邻域关系,例如KGNN。这些方法取得了可喜的结果,但很容易忽略药物分子的结构信息。
基于分子图的方法 药物分子可以通过以原子为节点、化学键为边的图自然地编码。 GNN的出现引发了对 DDI 预测中分子图表示的探索。特别是,专为图结构数据设计的图卷积网络(GCN)已广泛应用于药物研究中的空间特征提取。最近,识别对DDI预测贡献最大的关键子结构对于GNN来说是一个挑战。基于子结构的GNN旨在捕获基于分子中化学官能团的重要子结构,例如CASTER。基于分子图的方法在各种数据集上显示出有希望的性能。然而,这些方法仅将分子图或子结构视为固定大小,因此它们使用具有预定节点特征的 GNN 来捕获结构信息。
2.2 多视图学习
与上述以单一视角建模DDI任务的方法不同,多视图学习方法被提出以有效的模式结合两个或多个视角,例如GoGNN 和MUFFIN。近年来,对比学习已成功应用于基因调控相互作用和药物-靶点相互作用,但很少应用于DDI预测。例如,MIRACLE将DDI网络视为多视图图,将GCN与对比自监督相结合。 AMDE采用MPNN和Transformer分别学习药物的图特征和序列特征。与这些多视图学习模型不同,MVA-DDI引入了一种新颖的注意力网络,可以通过采用序列和图编码器来有效利用药物的不同视角来提高性能。
在深度学习用于 DDI 预测的研究中,ADME与我们的工作最相关。与ADME相比,该框架采用大规模数据集上的预训练策略,通过对比学习获得更泛化的GNN节点嵌入,而现有方法通常对节点嵌入进行初始化或自定义,缺乏良好的泛化能力。此外,我们的工作还提出了一种有效的自注意力机制来充分考虑序列和图特征的权重,因为从多角度获得的特征的不同维度对 DDI 预测有不同的影响。
3.方法
在本节中,我们将介绍拟议的 MVA-DDI 的技术细节。这里我们将 DDI 预测定义为二元分类任务。具体来说,给定药物对\(d_x\)和\(d_y\)(\(d_x, d_y \in D\)),其中\(D\)表示已知DDI的集合,我们的目标是学习预测函数 \(f : D \times D \times I\rightarrow\{0,1\}\)来推断概率这对是真正的药物与药物相互作用。
3.1 概述
MVA-DDI的整体框架如图1所示。首先,以药物SMILES序列作为输入,我们设计了两个特征提取模块,从SMILES序列和分子图中提取序列和图特征, 分别。具体来说,子序列由 ESPF 生成并馈入基于 Transformer 的序列编码器以获得序列表示。同时,以通过 RDKit 转换 SMILES 序列获得的分子图作为输入,利用基于 GCN 的图编码器来导出图表示。为了学习更多信息表示,引入了自我监督的预训练策略。其次,设计了一个自注意力特征聚合模块来获得集成的药物表示。最后,以综合药物表示作为输入,分类器输出 DDI 的相互作用概率。

图1. 所提出的MVA-DDI模型的总体架构。 (A) MVA-DDI 概述。 (B) 自监督模块。 (C) 特征聚合模块。
3.2 基于 Transformer 的序列编码器
我们设计了一个基于 Transformer 的序列编码器来提取 SMILES 序列的化学背景。Transformer 是一个合适的选择,因为它广泛用于自然语言处理中的序列编码。具体来说,我们采用 ESPF 算法来促进序列编码过程。该算法可以将 SMILES 序列分解为更小的子序列或原子符号。然后通过预定义的字典将这些子结构映射到相应的嵌入向量。这样我们就得到了词嵌入\(WE\)和位置嵌入\(PE\)作为Transformer模型的输入。
一般来说,Transformer的输入是SMILES序列的词嵌入\(WE\)和位置嵌入\(PE\)。与传统的 Transformer 分别取这两个向量不同,我们采用嵌入层将\(WE\)和\(PE\)相加生成相同维度大小的向量\(X\),然后将其送入 Transformer,Transformer 主要依靠多头自注意力和前馈神经网络。自注意力机制是 Transformer 模型的核心组成部分。具体来说,可以表述如下。
其中 \(Q,K\)和\(V\)分别表示查询向量、键向量和值向量,\(W\)表示由神经网络初始化的相应权重,\(d_K\)表示特征向量\(K\)的维度。我们获得每个位置的上下文表示 x通过对输入序列执行自注意力计算。前馈神经网络由两个全连接层和一个激活函数组成,用于执行非线性变换并映射每个位置的表示。最后,得到序列特征\(F_s\)如下。
3.3 基于GCN的图编码器
| Atom Feature | Size | Description |
|---|---|---|
| Atomic symbol | 44 | [C, N, O, S, F, Si, P, Cl, Br, Mg, Na, Ca, Fe, As, Al, I, B, V, K, Tl, Yb, Sb, Sn, Ag, Pd, Co, Se, Ti, Zn, H, Li, Ge, Cu, Au, Ni, Cd, In, Mn, Zr, Cr, Pt, Hg, Pb, Unknow] (One-hot) |
| Atomic degrees | 11 | Degree of atoms in a drug [0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10] (One-hot) |
| Implicit value | 7 | Implicit valence of atoms [0, 1, 2, 3, 4, 5, 6] |
| Formal charge | 1 | The formal charge of the atom, which usually ranges from -3 to +3 |
| Radical electrons | 1 | The number of free radical electrons of an atom, which usually ranges from 0 to 2 |
| Hybridization | 5 | The atomic hybridization mode [SP, SP2, SP3, SP3D, SP3D2] (One-hot) |
| Atomic aromaticity | 1 | Whether the atom is aromatic or not [0/1] |
| Total hydrogen atoms | 5 | Total number of hydrogen atoms in the atom [0, 1, 2, 3, 4] (One-hot) |
表1:预定义原子特征列表。
我们设计了一个基于GCN的图编码器来提取分子图的结构特征。在这里,我们使用图表来模拟原子及其化学键之间的关系。GCN可以有效地编码图的结构信息。我们将特征矩阵和邻接矩阵分别表示为\(X \in R^{N \times d}\) 和 \(A \in R^{N \times N}\),其中\(d\)和\(N\)表示特征的维度和节点数。每个条目\(A_{i,j}\)表示节点\(i\)和\(j\)之间是否存在连接。以分子图为输入,编码器通过平滑邻居的特征来输出图表示。具体来说,如图1所示,我们首先使用RDKit将SMILES序列转换为分子图的二维结构,其中节点和边表示原子及其之间的关系。定义每个原子的特征向量,如表1所示。GCN的具体传播如下:
其中\(H^{(l)}\)表示第l层的特征矩阵,我们将\(H^0\)设置为\(X\)。为了结合\(A\)中所有节点的自特征,我们有\(\hat{A} = A+I\),\(I\)是恒等式矩阵,\(D\)为\(A\)的度矩阵,且\(\tilde{D} = \sum\tilde{A_{i,j}}\)。\(σ\)是非线性激活函数,例如\(ReLU\)。最后一层使用\(softmax\)进行分类预测。\(W(l)\)是当前层卷积变换的可训练参数矩阵。GCN中的卷积是根据邻接矩阵和特征向量的乘积来计算的。此操作允许每个节点与其相邻节点传播信息,从而更新每个节点的特征表示。最后,我们从最后一层获得图特征\(F_g\)向量,如下所示。
3.4 自监督预训练
本文的关键贡献在于自监督预训练模块的设计。它由数据预处理、数据论证和对比损失函数组成。
数据预处理。为了像训练前一样充分考虑药物的特性,我们从 ChEMBL(第 32 版)数据库中提取了 4,194 个 FDA 批准的小分子药物和 5,805 个处于临床前阶段的候选分子。同时,我们从DrugBank数据库中下载了1,704个药物,总共获得了11,703个小分子药物。然后我们去掉没有SMILES、重复的、RDKit无法解析的小分子,最终得到总共8722个小分子药物用于预训练。
数据论证。为了便于对比学习,需要获取更多的样本。数据论证方法旨在生成给定 SMILES 的相应正样本和负样本。该方法基于两种特定于图的操作,包括随机节点丢失和边缘修改,这会导致图特征的随机扰动并帮助模型学习附加信息。具体来说,正样本对是通过对当前 SMILES 实施 5 个随机节点 dropout 和 5 个随机边修改来生成的。具体操作是在节点特征矩阵中随机选取5列数据置为0,在邻接矩阵中随机选取5对数据进行值(即0或1)交换(类似于删除和删除)边缘的增加)。然后与自己组成正样本对,标记为1。负样本对是通过随机选择与当前SMILES不同的样本进行5次随机节点dropout和5次随机边修改得到负样本对,则负样本对为形成并标记为0,负样本的上述操作重复5次。
对比损失函数。基于 GCN 的对比自监督模块旨在增强药物特征的判别性表示。所提出的模型通过使用对比损失进行优化。对于一对样本(\(d_i, d_j\)),它们分配的标签用\(y_{ij} (y_{ij} \in \{0,1\})\)表示,其中 \(y_{ij} = 0\) 表示它们属于负样本对,否则相反为正样本对。我们将它们对应的输出向量分别表示为\(o_i\)和\(o_j\),它们之间的对比损失定义如下:
其中\(d(o_i,o_j)\)表示两个向量之间的距离,并且我们有\(d(o_i,o_j) = \| o_i − o_j \|\)。\(m\)是控制样本对之间距离的裕度参数。第一项和第二项分别对应于负样本对和正样本对的损失。最终的对比损失函数是通过将每个样本对的损失求和平均得到的,如下所示:
其中\(N\)代表样本对的数量,我们有\(L^{i}_{cl} = L_{ij}\)。对比自监督预训练模型的整个传播过程如下。在前向传播过程中,输入是输出向量\(o_i\)和\(o_j\),以及它们的标签\(y_{ij}\)。前馈函数首先计算两个向量之间的\(L_2\)距离,称为欧氏距离。然后根据公式计算对比损失。最后,将对比损失作为反馈信号返回,用于反向传播以优化网络参数。
3.5 特征聚合
为了获得药物表示的最终嵌入,采用有效的特征聚合模块将序列和图特征向量组合成更具表现力的特征向量。对于序列特征\(F_s\)和图特征\(F_g\),设计了如下三种特征聚合方法。
相加。这是获得药物特征最终表示的简单而有效的方法。该操作是通过将两种类型的特征逐元素相加来实现的。计算公式为:
拼接。拼接操作是通过沿某个轴拼接多个特征来实现的。该方法保留了各个特征的所有信息,特征向量维度的增加可能会影响模型的复杂性。
注意力。注意力操作是一种基于加权平均的方法。它涉及根据不同特征向量的重要性为其分配权重,然后通过加权和将它们组合起来以获得最终的特征向量。这种方法允许动态调整分配给每个特征向量的权重W,这可以通过基于相关性强调或弱化特定特征来实现更精细的融合效果。
3.6 药物相互作用预测
给定一个DDI元组\((d_x,d_y,r)\),DDI预测可以表示为联合概率如下:
其中\(\sigma\)是sigmoid函数,\(W_{xy}\)和\(U_r\)是交互类型\(r\)的可学习表示,并且\(\odot\)表示本工作中使用的注意力特征聚合。所提出的MVA-DDI模型的学习过程可以通过最小化交叉熵损失函数来实现,如下所示。
反向传播从输出层传播到每个前面的层。端到端方法用于训练模型中的所有可训练参数。
4.结果
在本节中,我们首先介绍实验设置,然后通过与基线方法进行比较来证明所提出的模型 MVA-DDI 的性能。
4.1 数据集
我们实验中使用的已知 DDI 对是从 DrugBank (v5.1.9) 数据库下载的,我们在 1,704 种已批准的小分子药物之间提取了 124,725 个药物-药物对。此外,我们还收集了这 1,704 种药物的 SMILES 序列和类别信息。
4.2 基线方法
为了验证模型的性能,我们将 MVA-DDI 与相同 DrugBank 数据集上的基线方法进行比较。为了公平比较,所有基线方法都使用默认参数。接下来,我们简单介绍每种方法如下:
- DeepWalk是一种针对表示学习提出的基于图的方法。
- GraRep通过将图的全局结构信息集成到学习过程中来学习低维表示。
- GAE是一种图自动编码器模型,旨在学习图的低维表示。
- DeepDDI是一种为预测DDI 而开发的深度神经网络方法。
- CASTER是一种深度学习方法,可对药物的功能子结构进行编码以进行 DDI 预测。
- KGNN提出了一种知识图神经网络,通过结合先验知识图来预测DDI。
- AMDE提出了一种用于 DDI 预测的多视图深度学习,它同时对 SMILES 序列和原子图进行建模以学习药物表示。
4.3 指标
我们使用四个众所周知的测量指标来评估性能,即准确率(ACC)、ROC 曲线下面积(AUROC)、精确率-召回率曲线下面积(AUPR)和 F1 分数。
4.4 实验设置
在这项工作中,我们进行标准的 5 倍交叉验证(CV)来评估模型的性能。特别是,我们将已知的药物对随机分为五组。对于每一轮,我们依次选择一组药物对进行模型测试,而其余四组用于模型训练。预训练时,正负样本对的比例设置为1:5。对于序列编码器,我们将注意力头的数量设置为8。序列特征的长度设置为50。GCN编码器中的输出维度为75。我们使用Adam算法进行模型优化。学习率设置为 1e-4,训练周期为 50。
4.5 性能评估
在本节中,我们将 MVA-DDI 模型的性能与基线方法进行比较。表2显示了比较结果。可以看出,MVA-DDI 在四个指标方面始终优于基线方法。具体来说,MVA-DDI 的平均 ACC 为 94.49%,平均 AUROC 为 98.43%,平均 AUROC 为 98.45%,平均 F1 分数为 94.55%,比第二最佳方法 AMDE 提高了 2.57%、1.32分别为 %、1.68% 和 1.17%。此外,从表II中我们可以发现,多视图学习模型(即AMDE和MVA-DDI)比单视图模型(例如DeepDDI和KGNN)具有更好的性能,这表明从多个角度学习表示有助于提高模型的性能。模型的性能。虽然 MVA-DDI 和 AMDE 都使用分子序列和图形信息来学习表示,但与 AMDE 相比,MVA-DDI 有两个主要优点。首先,MVA-DDI 能够比 AMDE 学习更多信息丰富的图形表示。 MVA-DDI引入了预训练机制,使用大量先验药物数据而不是训练药物数据来学习图特征。其次,在整合不同视图的表示时,MVA-DDI 采用了一种注意力机制,允许模型自适应聚合特定于视图的表示,而 AMDE 使用简单的求和和串联方法。
| Model | ACC | AUROC | AUPR | F1 |
|---|---|---|---|---|
| DeepWalk | 83.44±0.07 | 91.76±0.04 | 90.64±0.05 | 83.53±0.08 |
| GraRep | 84.45±0.06 | 92.30±0.14 | 91.15±0.07 | 84.64±0.12 |
| GAE | 81.35±0.46 | 88.82±0.32 | 85.95±0.42 | 82.53±0.40 |
| DeepDDI | 82.81±0.21 | 88.38±0.53 | 89.23±0.84 | 83.53±0.21 |
| CASTER | 84.38±0.11 | 91.57±0.09 | 91.60±0.16 | 85.60±0.12 |
| KGNN | 89.53±0.16 | 93.64±0.27 | 92.86±0.28 | 90.06±0.11 |
| AMDE | 91.92±0.46 | 97.11±0.29 | 96.77±0.34 | 93.38±0.37 |
| MVA-DDI | 94.49±0.44 | 98.43±0.34 | 98.45±0.33 | 94.55±0.39 |
表2:MVA-DDI与基线之间的性能比较。
5.讨论和结论
在本文中,我们提出了一种新颖的可解释多视图注意力网络,称为 MVA-DDI,用于 DDI 预测。MVA-DDI 是一个双通道表示学习框架,充分利用 SMILES 序列和分子结构信息。特别是,对于 SMILES 序列,我们设计了一个基于变压器的编码器,通过考虑原子的化学背景来学习序列表示。对于分子图,我们使用自监督图对比学习编码器来学习图表示。这种自监督学习编码器允许模型通过对大量药物分子进行预训练来提取信息丰富的图形表示。为了有效地结合从两个视图获得的表示,引入了注意机制,通过考虑不同视图对每种药物的重要性来自适应地融合序列和图表示。大量实验结果表明,我们的 MVA-DDI 在预测药物相互作用方面优于七种最先进的方法。
![[React]AntDesign 4.x 汉化](https://img2024.cnblogs.com/blog/2281381/202412/2281381-20241216163539547-1399172913.png)