主要功能:
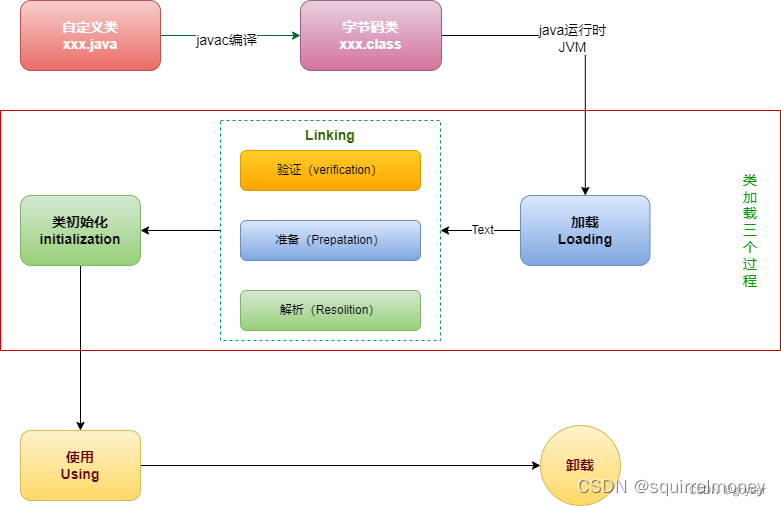
“开发了一个集成的用户界面C++程序,通过VASP代码进行高通量(HT)第一性原理计算,简称VASPMATE,具有强大的预处理功能,可用于各种结构建模和计算参数设置以及后处理分析用于电子、能量和其他特性。对于前者,包括等效晶胞的重新定义、坐标系的转换、原子坐标的修改和约束、超晶胞的构造和变形、 k点的设置以及各种必要的参数以及自动组合的潜力。后者旨在提取和分析原始数据以生成电子、物理和化学性质,例如 Kohn-Sham 轨道、能带结构、态密度、电荷密度差、费米面、热能校正、形成焓等。此外,VASPMATE提供了简单而整洁的命令模式,可以轻松地使用轻量级脚本构建多个健壮的HT工作流程。它还在 SPaMD 中实现,以方便用户友好的图形界面的设计,这有助于定制复杂的工作流程,以通过第一原理计算自动推导各种属性。特别是,SPaMD中提供了大量的自动例程,通过它可以向服务器提交一批HT任务,并提取计算结果并生成统计报告。通过进行多次评估和测试,该程序的效率和功能得到了严格的验证,这为其有针对性的第一原理计算的潜在应用提供了指导和信心。我们相信该程序将对计算材料科学领域的用户有所帮助。”——取自文章摘要。
Figure 2

Figure 3
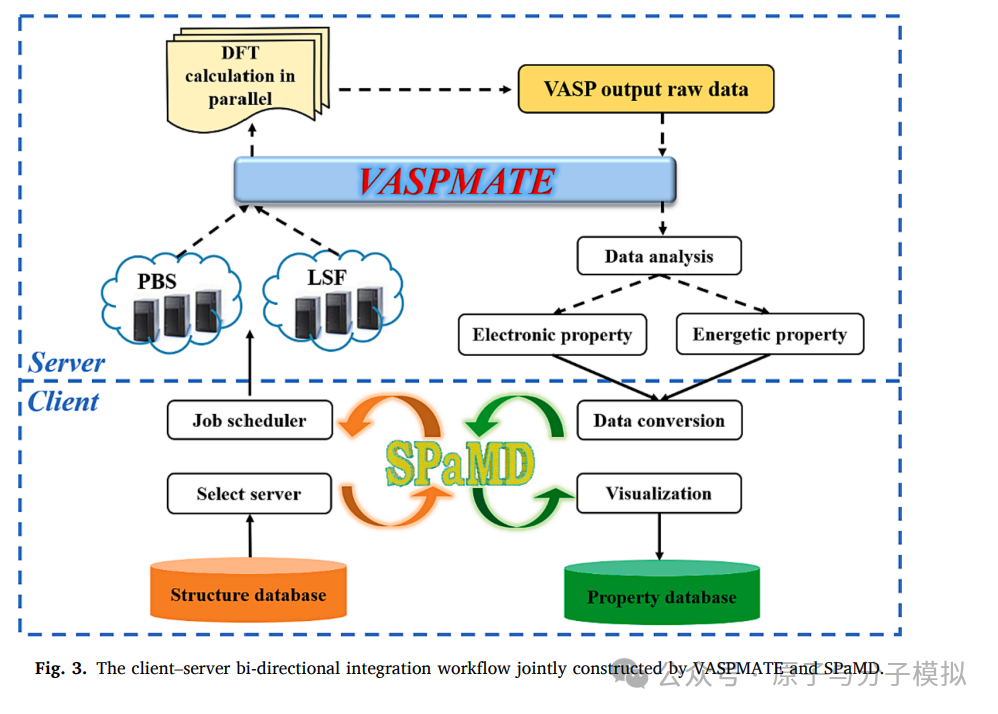
Figure 4
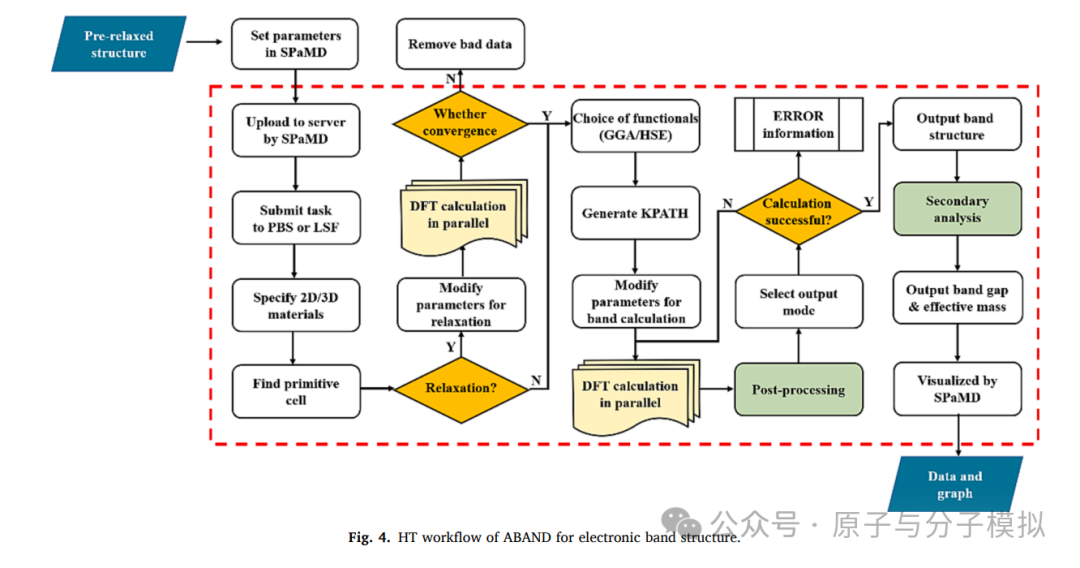
Figure 5
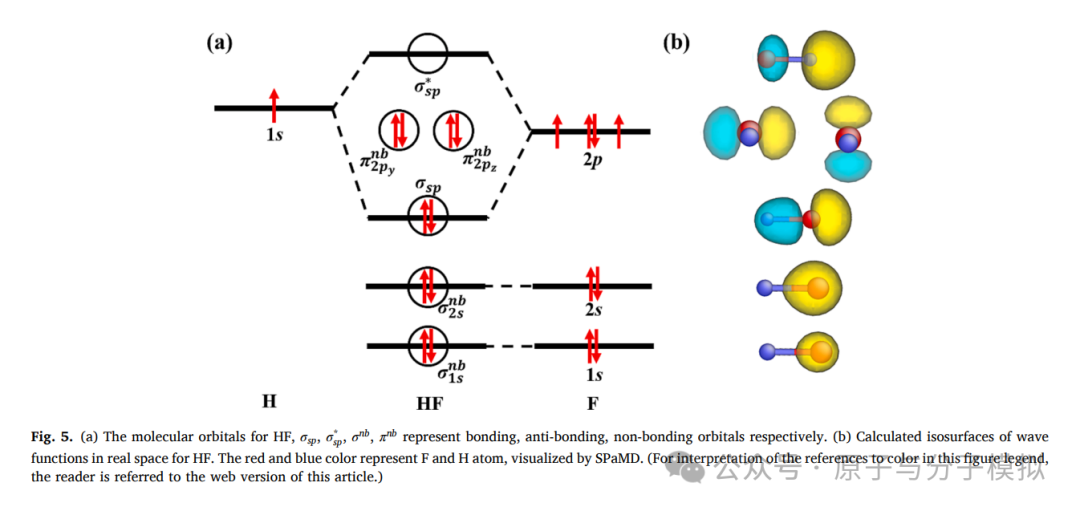
Figure 6
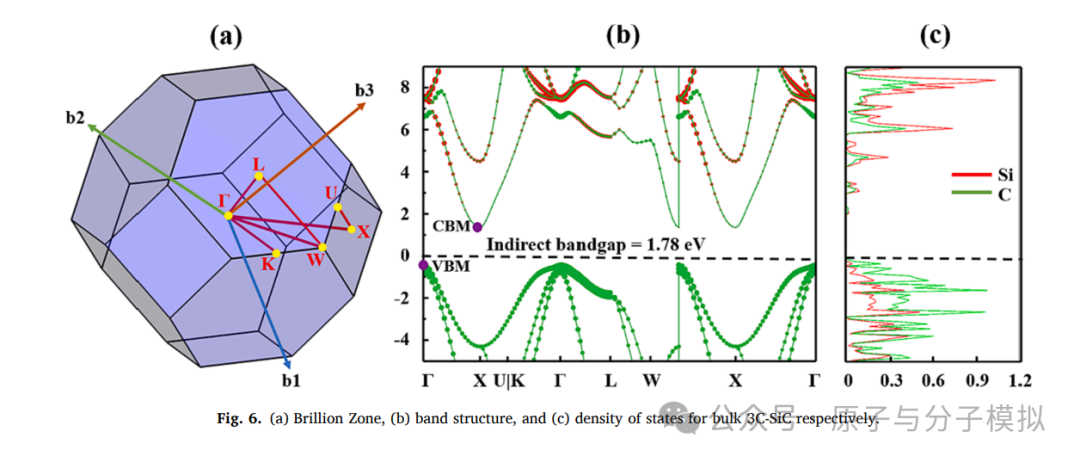
Figure 7
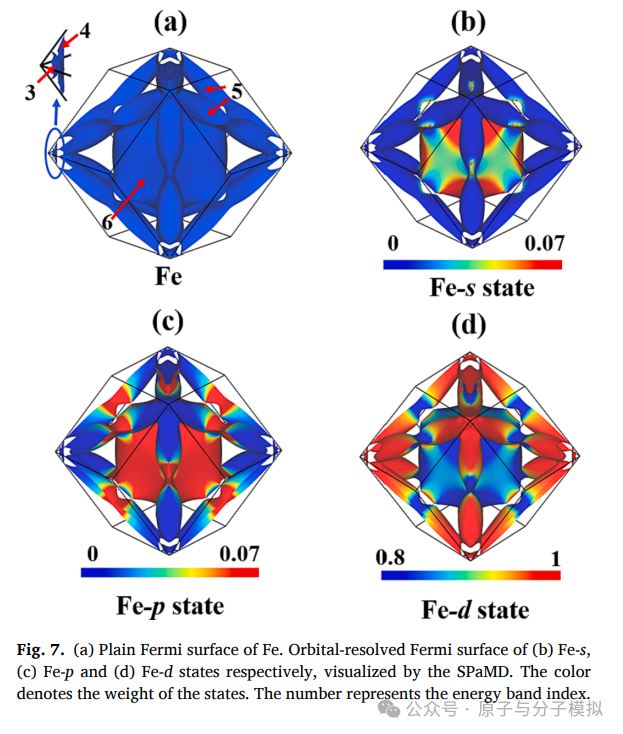
Figure 8
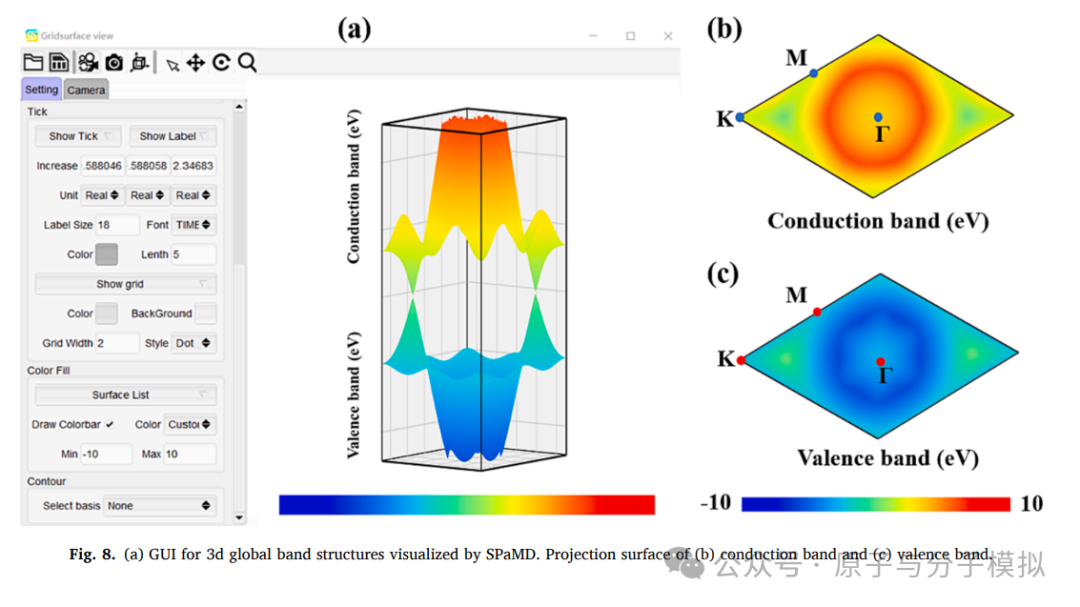
Figure 9
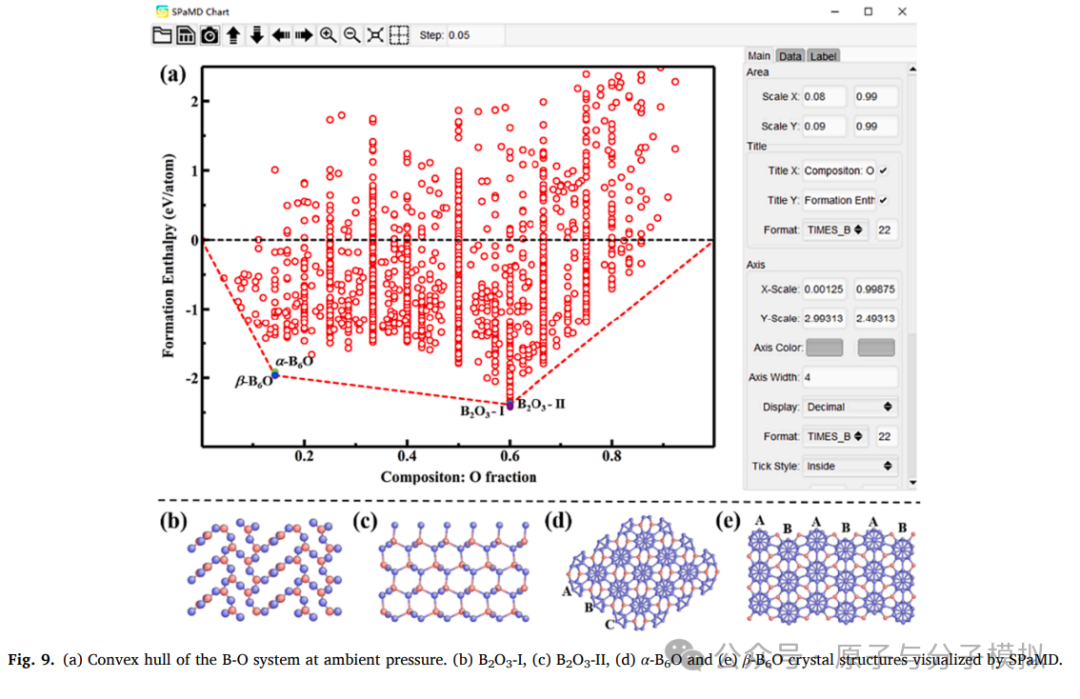
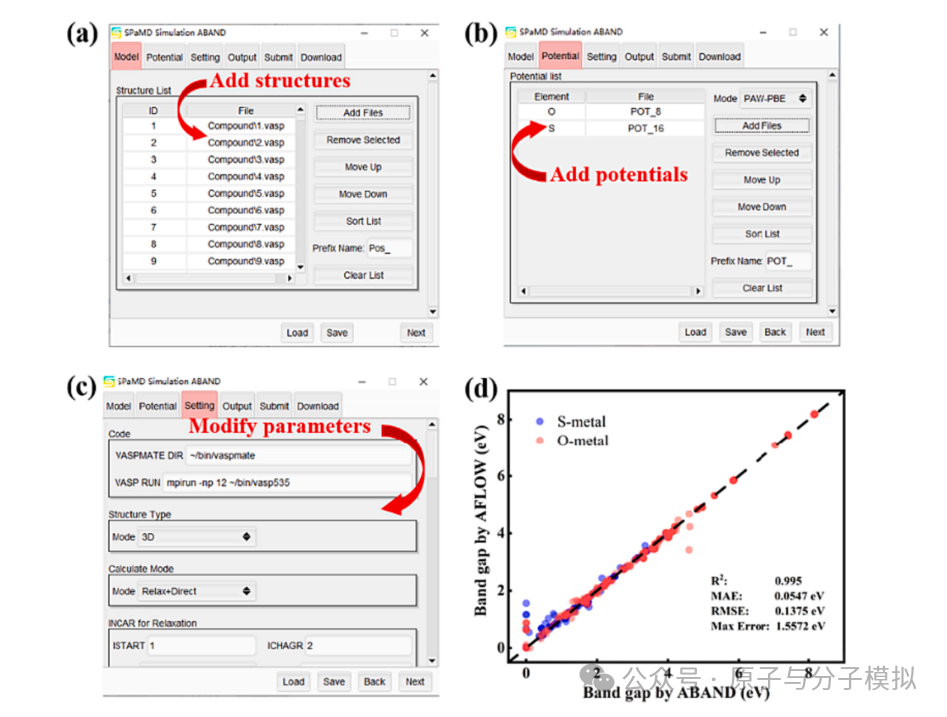
Figure 10
================================
![]()
以上是我们分享的一些经验或者文章的搬运,或有不足,欢迎大家指出!
如有侵权,请联系我立马删除!
详细内容(文章题目、文章链接、附件下载)可在微 信 公 众 号:原子与分子模拟获取,欢迎大家关注。