实验篇——家族成员染色体位置分析
文章目录
- 前言
- 一、名词解释
- 二、实操
- 1. 获取存储基因ID的文件
- 2. 获取基因密度文件
- 3. 获取染色体文件
- 4. 执行
- 总结
前言
在基因家族分析中,通过观察基因家族成员在染色体上的位置。可以判断在染色体上是否成簇分布。
一、名词解释
染色体位置分析是一种用于研究基因在染色体上具体位点的方法。
在家族成员染色体位置分析中,我们会标注出家族成员之间共享的染色体位点,观察基因家族成员在染色体上是否成簇分布。
如果基因家族成员在染色体上的位置分散、随机分布,则说明其不成簇分布,反之则是
二、实操
基于TBtools软件
点击菜单栏中的"Graphics",再点击 “Show Genes on Chromosome”,最后点击 “Gene Location Visualize from GTF/GFF”.
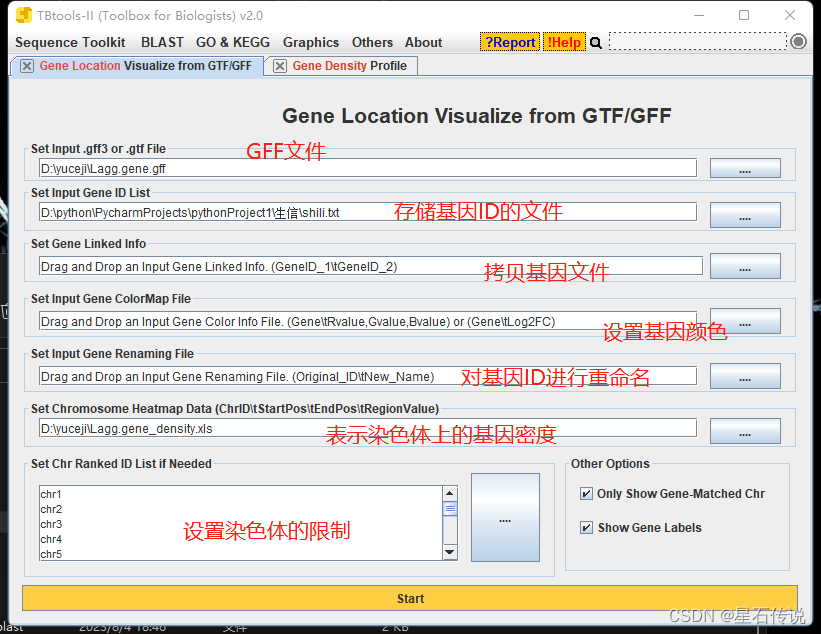
从上往下,第一和第二个空是必填的。其余可选填。
1. 获取存储基因ID的文件
获取存储基因ID的文件(获取的基因ID要与gff文件中的对应)
def quchu_geneid(gene_file, output_file):with open(gene_file, "r") as f:sequences = f.read().split(">")[1:]gene_ids = []for sequence in sequences:lines = sequence.split("\n")gene_id = lines[0].split(" ")[0]gene_ids.append(gene_id)with open(output_file, "w") as f:for gene_id in gene_ids:f.write(gene_id + "\n")gene_file = "D:\yuceji\Lindera_aggregata.gene.pep"
output_file = "gene_ids.txt"
quchu_geneid(gene_file,output_file)或者使用Linux中的sec工具:
sed -n 's/^>\([^ ]*\).*/\1/p' /home/wuyao.pep >output
less -SN output
前面两种方法都是直接基于pep文件提取基因ID,我们也可以从gff文件中提取。具体代码请看另一篇文章:
鉴定不同基因的复制模式
在这篇文章中的对gff文件修改的代码中。我们可以只提取基因ID,简单修改一下代码就能使用了。
2. 获取基因密度文件
获取基因密度文件
点击菜单栏中的"Sequence Toolkit",再点击 “GFF3/GTF Manipulate”,最后点击 “Gene Density Profile”.
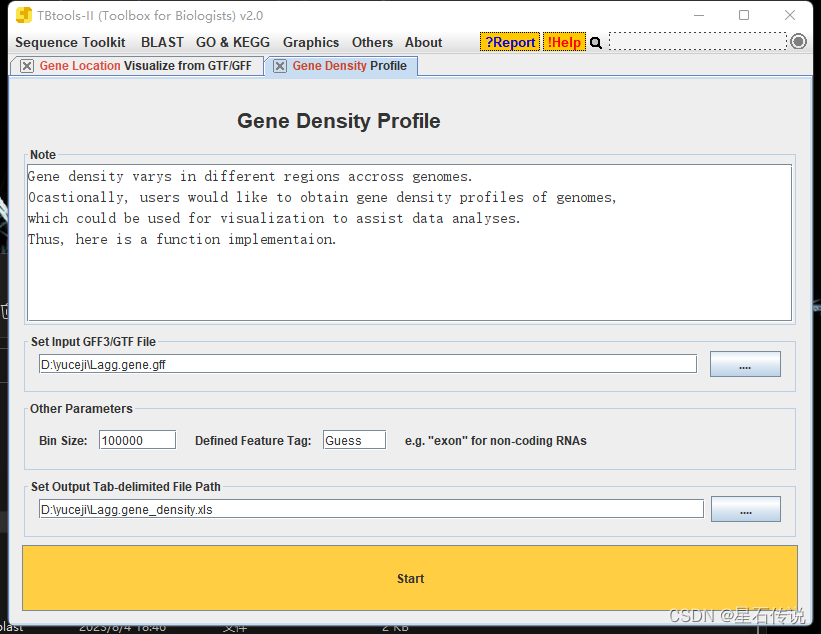
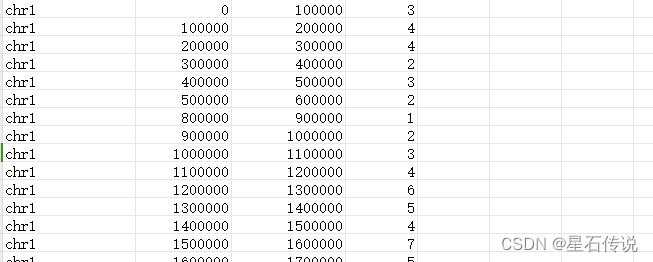
在给定的基因密度文件中,这四列分别代表以下内容:
第一列:染色体名称。它指示了每个区域所属的染色体。
第二列:起始位置。它表示每个区域的起始位置(以碱基为单位)。
第三列:终止位置。它表示每个区域的终止位置(以碱基为单位)。
第四列:基因密度。它表示在每个区域内检测到的基因数目或相关的基因特征的数量。
3. 获取染色体文件
获取染色体文件,从gff文件中提取
import pandas as pddef quchu_chr(gff_file, output_file):data = pd.read_csv(gff_file, sep="\t", skiprows=1)data = pd.DataFrame(data)chr_col = data.iloc[:, 0].unique()chr_col = pd.Series(chr_col)chr_col.to_csv(output_file, index=False,header= False)quchu_chr("D:\yuceji\Lagg.gene.gff", "chr_output.csv")
4. 执行
示例执行(随便提取的基因ID):
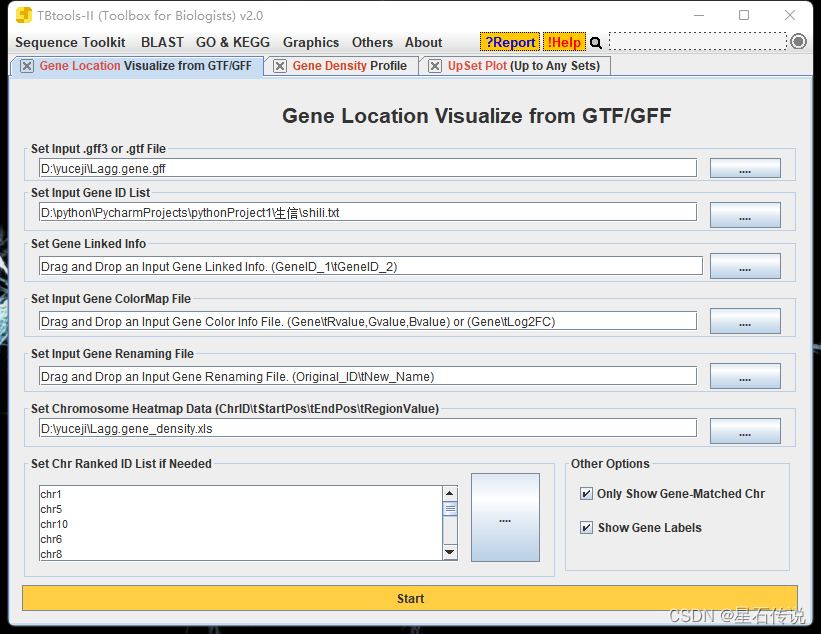
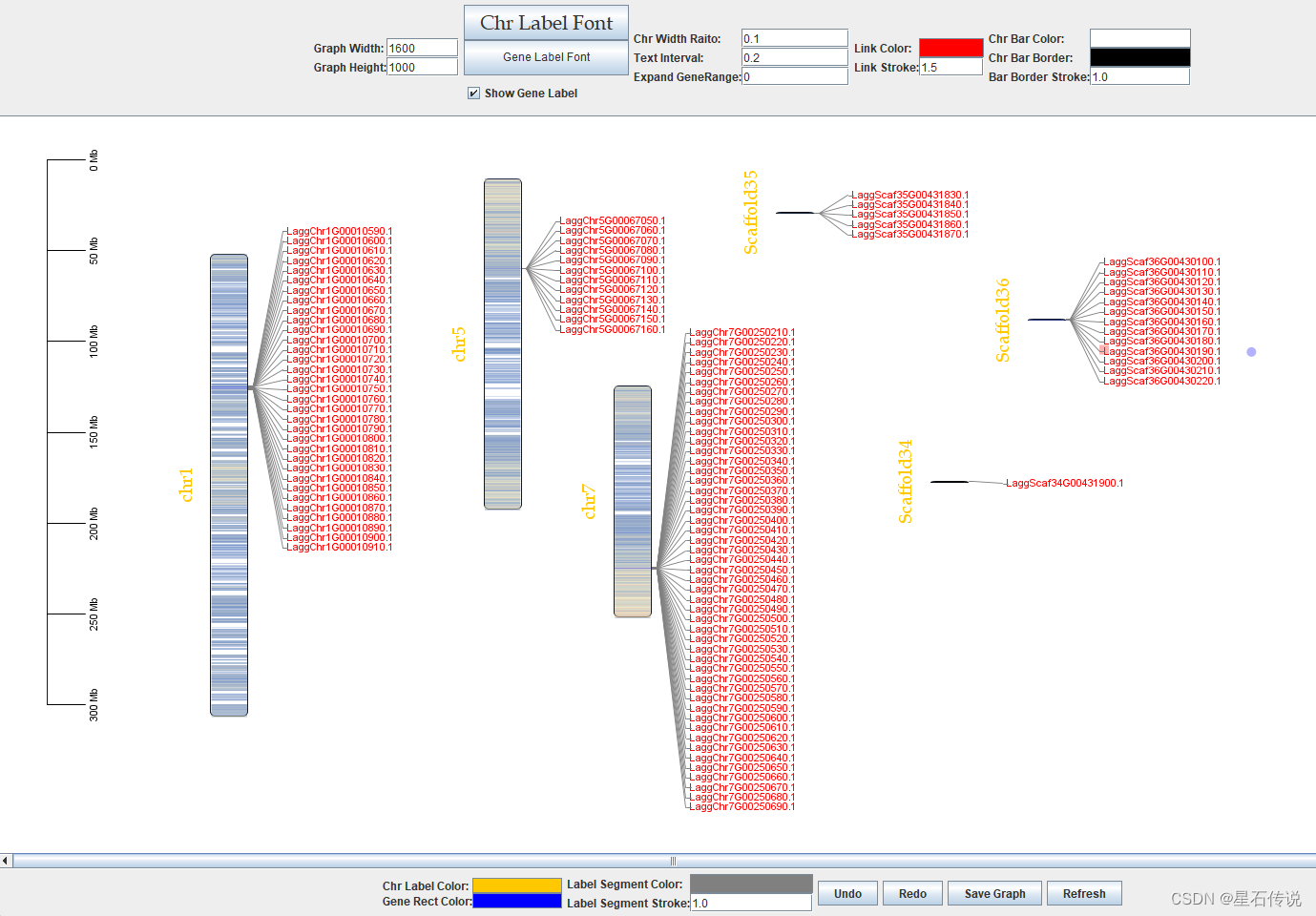
如果希望了解家族成员基因在染色体上的位置,观察是否成簇分布,可以输入包含家族成员基因ID的文件。
总结
本章是在TBtools软件上进行操作的,十分快速得得到了染色体位置分析的可视化图形。通过对图形的观察来判断家族成员基因在染色体上是否成簇分布。
天苍苍,野茫茫,风吹草低现牛羊。
–2023-8-22 实验篇